Primär amyloidos (AL-amyloidos) är en sällsynt sjukdom som innebär proteininlagring i ett eller flera organ. Den kliniska bilden kan variera kraftigt. Ungefär 80 procent av patienterna har en monoklonal lätt immunglobulinkedja i urinen.
Amyloidosengagemang av hjärtat drabbar ca 50 procent av patienterna, vilket är förenat med mycket kort överlevnad. Vanlig hjärtsviktsbehandling kan vara direkt olämplig. Behandlingen bör riktas mot att med diuretika och kemoterapi avlasta ödem.
Avsaknad av kända riskfaktorer för hjärtsvikt med normal eller låg amplitud av QRS-komplex på EKG och samtidig hypertrofi och restriktivt fyllnadsmönster på ekokardiografi bör väcka misstanke om hjärtamyloidos.
Amyloidos innebär extracellulär vävnadsdeposition av fibriller bestående av proteiner som strukturerar sig i »β-pleated sheets« och som är motståndskraftiga mot kroppens egna proteolytiska nedbrytningsmekanismer. Även om 25 olika amyloidgenetiska proteiner har beskrivits, är många av dem sällsynta och ger ingen hjärtpåverkan.
De tre vanligaste formerna som är definierade av sina prekursorproteiner är AL-amyloidos (primär [(A = amyloid, L = lätt immunglobulinkedja), AA-amyloidos (sekundär [protein AA]) och transtyretinderiverad amyloidos. Vid AL-amyloidos produceras en monoklonal lätt immunglobulinkedja av en plasmacellsklon i benmärgen. Monoklonalt immunglobulin i serum och/eller en monoklonal lätt immunglobulinkedja i urinen är påvisbar i 80 procent av fallen.
Klinisk evidens av kardiellt engagemang uppgår till ca 50 procent av alla patienter med AL-amyloidos [1] jämfört med endast 5 procent vid AA-amyloidos [2]. Vid transtyretinderiverad (TTR) amyloidos, som finns både i familjär form och som icke-ärftlig senil systemamyloidos, är mutationer i genen för TTR ofta associerade med amyloid kardiomyopati.
Fallbeskrivning
En tidigare frisk 51-årig man med hereditet för hjärt–kärlsjukdom – modern hade plötsligt gått bort i misstänkt hjärtinfarkt – sökte akutmottagningen i januari 2013 på grund av ansträngningskorrelerade centrala bröstsmärtor sedan sommaren 2012, vilka hade accentuerats. EKG visade sinusrytm med T-vågsnegativisering inferiolateralt utan tecken till vänsterkammarhypertrofi (normal QRS-amplitud) och platåformat troponin-T-mönster på omkring 30 ng/l.
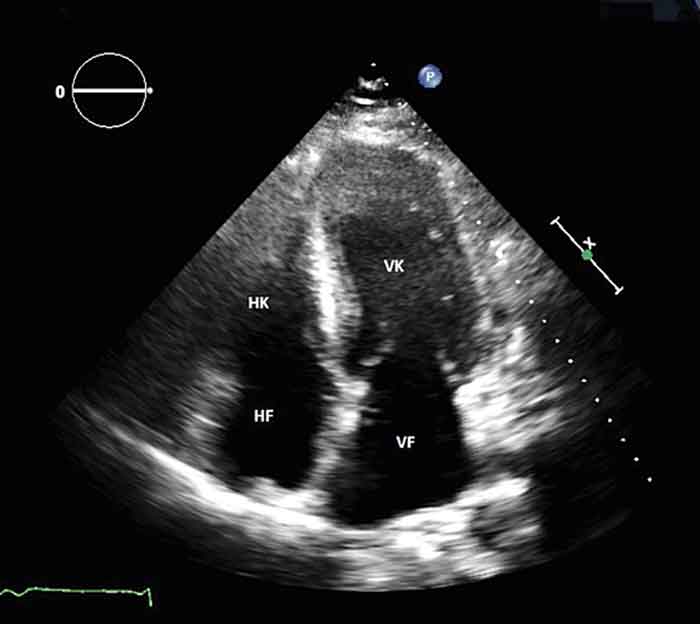
Patienten lades in på hjärtintensivvårdsavdelning, och en koronarangiografi genomfördes, vilken påvisade diskret ateromatos i LAD (främre nedåtstigande grenen av vänster koronarartär) och RCA (höger koronarartär) men inga signifikanta stenoser eller synliga tromber. Ekokardiografi visade måttligt hypertrof vänster kammare med normal systolisk funktion, bilateralt måttligt dilaterade förmak och diastolisk dysfunktion i det närmaste som restriktivt fyllnadsmönster (E/A-kvot = 1,5; decelarationstid 70 ms; IVRT (isovolumetrisk relaxation) 70 ms och lägre systoliskt lungvenstryck än diastoliskt [PVs < PVd]).
Under vårdtiden hade patienten två korta episoder av förmaksflimmer och ett genomsnittligt blodtryck på 135/65 mm Hg. Lipidstatus var diskret förhöjt utan tecken till ärftliga lipidstörningar med hänsyn till moderns befarade hjärtsjukdom. Han var sedan tidigare medicinfri och hade aldrig konstaterats ha högt blodtryck vid ordinarie kontroller på vårdcentral.
Han skrevs ut med atorvastatin 40 mg och metoprolol 100 mg.
Efter 2 veckor återkom patienten till akutmottagningen med fortsatta episoder av tryckkänsla centralt i bröstet samt dyspné som förvärrades vid ansträngning. Han hade då förmaksflimmer med frekvens 125 med diskreta pittingödem, lätt troponinläckage utan dynamik och i övrigt normala laboratoriefynd.
Patienten lades in på arytmiavdelningen på kardiologklinik, och man titrerade upp dosen metoprolol till 200 mg. Hjärtrytmen slog om till sinusrytm, och han skrevs ut med planerat återbesök på arytmimottagningen.
Efter utskrivningen mådde patienten allt sämre, och symtomen förvärrades. I samband med besök på vårdcentralen sänktes betablockaden, och patienten började med jämna mellanrum utveckla febertoppar upp till 39–40 grader. Datortomografiundersökning av torax visade ca 6 cm pleuravätska på höger sida, mindre mängd ascites, lätt emfysem och lätt ökat antal normalstora körtlar i mediastinum. Torakocentes utfördes på lungmottagningen; inga maligna celler kunde påvisas. TB-odling togs.
Patienten återkom kort därefter med accentuerade besvär och erhöll pleuradränage; förnyad cytologi genomfördes. Hjärtsviktsmarkören NT-proBNP (N-terminal pro-brain natriuretic peptide) var på drygt 10 000 ng/l. Datortomografiundersökning efter uttappad vätska för kartläggning av pleura och parenkym visade normala fynd. Man misstänkte diastolisk hjärtsvikt som orsak till symtomen, och behandling med ramipril sattes in och ett snart återbesök planerades.
Vidare utredning på vårdcentral visade nyupptäckt M-komponent (monoklonalt immunglobulin) av IgA-typ på 8,9 g/l med sänkt bakgrundsgamma. Dessutom påvisades Bence Jones proteinuri med 315 mg/l kappakedjor. Ingen hyperkalcemi uppmättes, och man såg inte några skelettförändringar vid datortomografiundersökningen. Ytterligare en pleuratappning gjordes på lungmottagningen, och QuantiFERON-test utföll negativt.
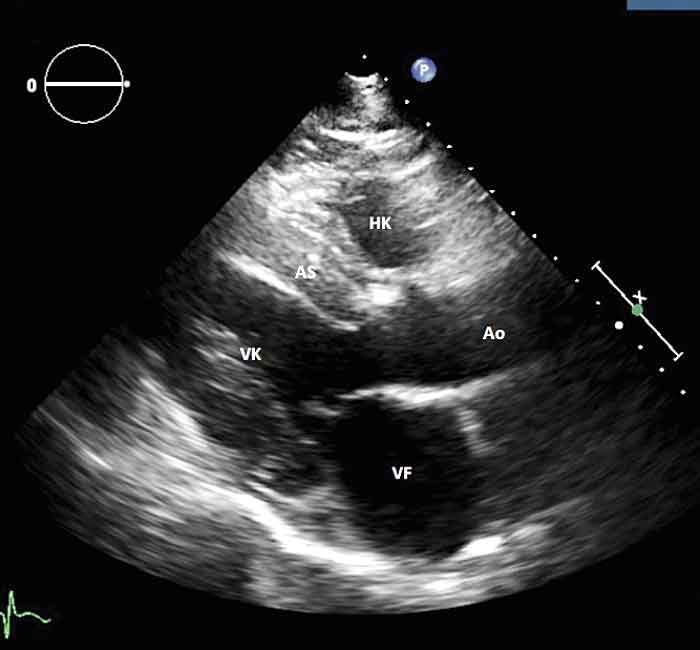
Patienten remitterades kort därefter till hjärtsviktsavdelning och genomgick en ny ekokardiografiundersökning, som denna gång visade kraftigt hypertrof vänsterkammare med lätt till måttligt sänkt funktion globalt, ejektionsfraktion 40–45 procent, kraftigt dilaterat vänster förmak och restriktivt fyllnadsmönster som progredierat (E/A-kvot = 2,4, PVs < PVd). Höger kammare var kraftigt dilaterad med lätt sänkt funktion, och att döma av gradienten över trikuspidalklaffen förelåg normalt estimerat tryck i pulmonalartären.
Vid närmare anamnes påtalade patienten domningar och känselbortfall i höger ben. Blodtrycket var nu lägre, 95/65 mm Hg. Misstanke om hjärtamyloidos väcktes, och undersökning med magnetisk resonanstomografi (MRT) av hjärtat utfördes. Denna visade biventrikulär hypertrofi med LVEF (vänsterkammarejektionsfraktion) på ca 40 procent och kraftigt nedsatt rörlighet i atrioventrikulärplanet. Viabilitetssekvenser visade mönster som kan ses vid inlagringssjukdom.
Bukfettsbiopsi med specialfärgning med alkalisk kongo påvisade amyloidinlagring. Under vårdtiden gjordes en benmärgsbiopsi, som visade rikligt med avvikande plasmaceller.
Pleurodesbehandling med snar uppföljning på hematolog- och hjärtsviktsmottagning planerades. Patienten lades kort därefter in för nytt pleuradränage samt pleurodes men drabbades av hjärtstillestånd under vårdtiden. Trots långvarigt återupplivningsförsök avled patienten. Eventuell hematologisk behandling hann aldrig påbörjas.
Diskussion
AL-amyloidos är en sällsynt förekommande sjukdom där den exakta incidensen är okänd. I USA uppskattar man den till 6–10 fall per miljon personår [3]. AL-amyloidos debuterar oftast hos personer >40 års ålder [4] och är i allmänhet systemisk. Prognosen är till stor del beroende av vilka organ som har engagerats. När hjärtat är engagerat är prognosen hos en obehandlad patient mycket dålig. Transtyretinderiverad amyloidos är oftare associerad med lindrigare kliniska manifestationer, långsammare progress och bättre prognos [5]. Skillnaden i mortaliteten har demonstrerats hos patienter med hjärtamyloidos med liknande ekokardiografiska förhållanden: 1-årsöverlevnaden var 92 procent hos patienter med den familjära formen och 38 procent vid AL-amyloidos [6].
Systemisk amyloidos ska misstänkas framför allt vid nefros hos icke-diabetiker, kardiomyopati i avsaknad av hypertoni eller ischemisk hjärtsjukdom, hepatomegali, perifer eller autonom neuropati, purpura i ansikts-/halsregion och makroglossi. AL-amyloidos kan förekomma som ett sekundärt fenomen vid myelom, mer sällan vid Waldenströms sjukdom.
Förekomst av M-komponent är ett relativt vanligt fynd i befolkningen; prevalensen tros vara omkring 1 procent och stiger kraftigt med ökad ålder, hos personer >50 år är prevalensen omkring 3 procent [7]. I en Malmö-studie bland 930 patienter med nyupptäckta M-komponenter under perioden 1975–1989 noterade man den primära diagnosen »monoklonal gammopati av oklar signifikans« (MGUS) hos 72 procent, myelom 19 procent, Waldenströms sjukdom 2 procent och annan lymfoproliferativ sjukdom hos 6 procent, endast 1 procent av patienterna fick diagnosen amyloidos [8].
Biopsi ger säkrast diagnos
Den säkraste metoden för diagnostisering av amyloidos är biopsi av misstänkt engagerat organ eller (vanligast) bukfettsbiopsi för amyloidfärgning och typning av amyloid. Plasma- och urinelektrofores, benmärgsprov med frågeställning om monoklonala plasmaceller, kvot av fria lätta immunglobulinkedjor (FLC) och blodbild är andra undersökningar som ingår i utredningen [9].
När amyloidos är konstaterad bör man typbestämma amyloidinlagringen biokemiskt. Analys och amyloiddiagnostik genomförs på bl a Akademiska sjukhuset i Uppsala med framför allt en metod som bygger på Western blot-teknik (antikroppar) [10]. Immunhistokemisk analys finns också tillgänglig men är förknippad med vissa svårigheter, eftersom kommersiellt tillgängliga antikroppar ofta fungerar dåligt för amyloidtypning. Exakt biokemisk typbestämning är nödvändig för rätt behandling.
Högersidiga hjärtsviktssymtom vanligt vid hjärtamyloidos
De vanligaste orsakerna till diastolisk hjärtsvikt är långvarig hypertoni, kranskärlssjukdom, hypertrof kardiomyopati, klaffsjukdom, diabetisk hjärtsjukdom, stigande ålder och restriktiv hjärtsjukdom som innefattar både hereditära och icke-hereditära former (Fakta 1) [11]. Fysiologin vid restriktiv kardiomyopati kännetecknas av betydande styvhet i hjärtmuskeln med försämrad kammarfyllnad. Ekokardiografiskt ses oftast dilatation av förmaken och normal eller liten vänsterkammare med välbevarad systolisk funktion samt diastolisk dysfunktion med restriktivt mönster. Väggtjockleken kan dock vara ökad vid vissa infiltrativa tillstånd som amyloidos och Fabrys sjukdom.
Vid hjärtamyloidos är högersidiga hjärtsviktssymtom som perifera ödem, hepatomegali och ascites vanliga. Trots att trycket i vänsterhjärtat är ökat, är lungödem ovanligt. Ekokardiografiskt ses vänsterkammarhypertrofi med diastolisk störning initialt och ökade fyllnadstryck. Senare i förloppet kan man påvisa förstorade och immobila förmak och icke-dilaterad vänsterkammare med systolisk dysfunktion. Restriktiv bild ses i mer avancerade fall sekundärt till att vänsterkammaren inte kan dilatera sig kompensatoriskt såsom vid andra kardiomyopatier [12, 13].
Amyloidinlagringen resulterar även i ökad ekogenitet. Detta har beskrivits som ett granulärt och glittrande utseende av myokardiet vid ekokardiografi [14]. Dock är detta en okänslig indikator för amyloidos, eftersom endast en liten andel av patienterna (26–36 procent) uppvisar detta mönster, och det har begränsad specificitet (sannolikt <71–81 procent) [15].
Vänsterkammarförtjockningen som syns vid ekokardiografi misstolkas oftast som vänsterkammarhypertrofi. Hypertrofi på grund av amyloidinlagring ger till skillnad från de riktiga hypertrofierna minskade (låga voltage) eller normala QRS-komplex på EKG. Kombinationen av vänsterkammarförtjockning med låga voltage i EKG-mönstret är relativt specifikt för infiltrativa kardiomyopatier [14, 16]. En studie visade att kombinationen låga voltage och kammarseptum >1,98 cm har en sensitivitet på 72 procent och specificitet på 91 procent [14]. Andra tillstånd som kan ge små EKG-komplex, t ex tamponad, perikardvätska eller emfysem, är inte associerade med ökad hjärtmassa, medan tillstånd med ökad hjärtmassa, t ex hypertoni, oftast är associerade med normal eller ökad amplitud i QRS-komplexen [17].
NT-proBNP och troponin T är prognostiska markörer
Serum-NT-proBNP och troponin T har visat sig vara sensitiva markörer för hjärtengagemang och kan användas som prognostiska markörer hos patienter med AL-amyloidos [18]. De är dessutom oberoende prediktorer för överlevnad och kan användas för utvärdering av behandlingseffekt [19]. Ischemi i kranskärl och småkärl orsakade av amyloidinlagring kan vara en bidragande faktor till ökade nivåer av troponin i plasma. De epikardiella kärlen är oftast skonade från amyloidinlagring, men engagemang av intramurala kärl förekommer hos 90 procent av patienter med AL-amyloidos [20, 21]. Detta kan eventuellt förklara vår patients initiala symtom med bröstsmärtor, där koronarangiografi inte påvisade några signifikanta stenoser.
Grundbehandlingen är symtomlindrande
Behandling av symtomatisk hjärtamyloidos grundar sig på hjärtrelaterade symtom och specifik terapi mot bakomliggande sjukdom. Grundbehandlingen för patienter med hjärtamyloidos är loop-diuretika och aldosteronantagonister för att häva de kraftiga ödemen, dvs symtomlindrande behandling. Dessa läkemedel är väl tolererade och ger inte kraftig hypotonibenägenhet. Effekt och säkerhet av behandling med ACE-hämmare och angiotensinreceptorblockerare är okänt hos patienter med hjärtamyloidos. Klinisk erfarenhet visar att dessa läkemedel kan framkalla uttalad hypotoni, sannolikt på grund av att man avslöjar en subklinisk autonom neuropati.
Kalciumblockerare som verapamil och diltiazem har i små studier använts för att sänka puls och förbättra ventrikulär relaxation hos patienter med diastolisk dysfunktion. De är dock helt ineffektiva vid hjärtamyloidos, eftersom dysfunktionen beror på amyloidinlagring och inte myokardcellsdysfunktion [22, 23].
Inga säkra data avseende morbiditet eller mortalitet föreligger för betablockad, och denna behandling kan i själva verket förvärra symtomen hos patienter med hypotoni eller vars slagvolym är beroende av hjärtfrekvensen. Digoxin är ett annat olämpligt läkemedel, eftersom det binder till amyloidfibriller och kan orsaka risk för digitalisintoxikation [24].
Majoriteten av patienterna har även extrakardiellt engagemang av amyloidos och är därför i de flesta fall inte aktuella för hjärttransplantation, eftersom sjukdomen kan progrediera i andra organ och även engagera det transplanterade hjärtat.
Förekomsten av förmakstromber förefaller vara hög hos patienter med AL-amyloidos och hjärtengagemang. Fler patienter med AL-amyloidos än patienter med andra former av amyloidos har intrakardiella tromber (51 procent mot 16 procent; P < 0,001) och fler tromboemboliska händelser (26 procent mot 8 procent; P < 0,03) [25]. Tidig antikoagulantiabehandling kan därför reducera både morbiditet och mortalitet. Implantering av defibrillator (ICD) förefaller inte sänka mortaliteten, eftersom elektromekanisk dissociation föreligger; detta har också illustrerats i en liten kohort på 19 patienter med AL-amyloidos med hjärtamyloidos [26].
Autolog stamcellstransplantation har prövats
Behandling av och prognos för patienter med AL-amyloidos beror till stor del av vilka organ som är involverade. Vid hjärtsvikt är medianöverlevnaden endast 4–6 månader [1]. Dock kan man med tidigt insatt behandling med kemoterapi förlänga överlevnaden [27]. De vanligaste förekommande kemoterapierna är melfalan med dexametason, eller bortezomib, cyklofosfamid och dexametason, där målet är att reducera fria lätta immunglobulinkedjor med 90 procent i serum. Högdos melfalan och autolog stamcellstransplantation har visat goda resultat och längre överlevnadstid (>4 år) bland selekterade patienter med AL-amyloidos som tål denna behandling [28].
Tillgängligheten av hjärtmarkörer (NT-proBNP och troponiner) har inneburit ett framsteg för stratifiering av patienter i tidigt eller avancerat stadium av hjärtamyloidos och för att underlätta valet av behandling och predicera utfallet av autolog stamcellstransplantation [29, 30].
Enskild läkare/klinik bör leda utredningen
Patienten i den aktuella fallbeskrivningen debuterade med kardiopulmonella symtom redan sommaren 2012 och dröjde ett halvår innan han sökte sjukvård. Dessvärre försenade läkarfördröjning diagnostiken ytterligare ett halvår innan misstanke om amyloid kardiomyopati väcktes.
AL-amyloidos är i allmänhet systemisk, och de kliniska konsekvenserna beror till stor del av vilka organ som är engagerade, vilket innebär att misstanke och eventuell utredning kan förekomma inom många specialiteter. Dessvärre upptäcks AL-amyloidos ofta sent i sjukdomsförloppet och är förenad med dålig prognos vid diagnos.
Denna fallbeskrivning illustrerar att utredningsgången försvåras av olika klinikers (inklusive primärvårdens) samtidiga arbete utan att någon enskild läkare leder den primära utredningen. Utredningen fördelades mellan kardiolog- och lungkliniken med ett antal besök på vårdcentralen däremellan på grund av försämring och nytillkomna symtom. Konsekvensen av detta kan vara fördröjning av diagnos och eventuell behandling. Vi rekommenderar därför att en enskild läkare/klinik, beroende på organengagemang och symtom, driver utredningen med hjälp av andra berörda specialiteter inklusive hematologer.
Fallbeskrivningen visar också att låga voltage i EKG-mönstret inte alltid förekommer hos patienter med hjärtamyloidos. Det restriktiva fyllnadsmönstret vid den första ekokardiografiundersökningen med avsaknad av hypertrofi på EKG, normalt blodtryck på avdelningen och normalt fynd vid cytologisk analys av pleuravätskan borde ha väckt misstanke om inlagringssjukdom tidigare. I efterhand kan man redan på den första ekokardiografin skönja ett glittrande utseende hos myokardiet (Figur 1).
Det är viktigt att tidigt ha inlagringssjukdom såsom hjärtamyloidos med i differentialdiagnostiska överväganden hos patienter med hjärtsvikt med bevarad vänsterkammarfunktion samt vänsterkammarhypertrofi utan tecken till hypertoni eller ischemisk hjärtsjukdom, oavsett om QRS-komplexet är normalt eller uppvisar låga voltage.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1.
Olika tillstånd som kan ge restriktiv kardiomyopati
Hereditära
- Hereditär (okänd gen)
- Sarkomerproteinmutationer
– troponin I-genmutation (restriktiv kardiomyopati ± hypertrof kardiomyopati)
– mutation i essentiella lätta myosinkedjor
- Hereditär amyloidos
- Transtyretin (restriktiv kardiomyopati + neuropati)
- Apolipoprotein (restriktiv kardiomyopati + nefropati)
- Desminopati
- Pseudoxanthoma elasticum (Grönblad–Strandbergs syndrom)
- Hemokromatos
- Fabrys sjukdom
- Glykogeninlagringssjukdomar
Icke-hereditära
- Amyloidos (AL)
- Sklerodermi
- Endomyokardiell fibros
– hypereosinofilt syndrom
– idiopatisk fibros
– kromosomorsakad fibros
– läkemedelsorsakad fibros
- Karcinoid hjärtsjukdom
- Metastaser
- Strålning
- Läkemedelsorsakad restriktiv kardiomyopati (antracykliner)
Referenser
- Dubrey SW, Cha K, Anderson J, et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM. 1998;91:141-57.
- Dubrey SW, Cha K, Simms RW, et al. Electrocardiography and Doppler echocardiography in secondary (AA) amyloidosis. Am J Cardiol. 1996;77:313-5.
- Kyle R, Linos A, Beard C, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-22.
- Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203-12.
- Desai HV, Aronow WS, Peterson SJ, et al. Cardiac amyloidosis: approaches to diagnosis and management. Cardiol Rev. 2010;18:1-11.
- Dubrey SW, Cha K, Skinner M, et al. Familial and primary (AL) cardiac amyloidosis: echocardiographically similar diseases with distinctly different clinical out-comes. Heart. 1997;78:74-82.
- Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362-9.
- Bird J, Behrens J, Westin J, et al; Haemato-oncology Task Force of the British Committee for Standards in Haematology, UK Myeloma Forum and Nordic Myeloma Study Group. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol. 2009;147:22-42.
- Systemisk immunoglobulin light chain (AL) amyloidos. Riktlinjer fastställda av Diagnosgruppen för plasmacellssjukdomar 2014-04-28 [citerat 13 dec 2014]. www.sfhem.se/Files.aspx?f_id=103289
- Westermark P, Davey E, Lindbom K, et al. Subcutaneous fat tissue for diagnosis and studies of systemic amyloidosis. Acta Histochem. 2006;108:209-13.
- Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-6.
- Patel AR, Dubrey SW, Mendes LA, et al. Right ventricular dilation in primary amyloidosis: an independent predictor of survival. Am J Cardiol. 1997;80:486-92.
- Klein AL, Hatle LK, Taliercio CP, et al. Serial Doppler echocardiographic follow-up of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol. 1990;16:1135-41.
- Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43:410-5.
- Selvanayagam JB, Hawkins PN, Paul B, et al. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol. 2007;50:2101-10.
- Simons M, Isner JM. Assessment of relative sensitivities of noninvasive tests for cardiac amyloidosis in documented cardiac amyloidosis. Am J Cardiol. 1992;69:425-7.
- Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol. 1982;49:9-13.
- Palladini G, Campana C, Klersy C, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107:2440-5.
- Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22:3751-7.
- Crotty TB, Li CY, Edwards WD, et al. Amyloidosis and endomyocardial biopsy: Correlation of extent and pattern of deposition with amyloid immunophenotype in 100 cases. Cardiovasc Pathol. 1995:4:39-42.
- Smith TJ, Kyle RA, Lie JT. Clinical significance of histopathologic patterns of cardiac amyloidosis. Mayo Clin Proc. 1984;59:547-55.
- Gertz MA, Skinner M, Connors LH, et al. Selective binding of nifedipine to amyloid fibrils. Am J Cardiol. 1985;55:1646.
- Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest. 1993;104:618-20.
- Griffiths BE, Hughes P, Dowdle R, et al. Cardiac amyloidosis with asymmetrical septal hypertrophy and deterioration after nifedipine. Thorax. 1982;37:711-2.
- Feng D, Edwards WD, Oh JK, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116:2420-6.
- Kristen AV, Dengler TJ, Hegenbart U, et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm. 2008;5:235-40.
- Lachmann HJ, Gallimore R, Gillmore JD, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol. 2003;122:78-84.
- Skinner M, Sanchorawala V, Seldin DC, et al. High-Dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
- Madan S, Kumar SK, Dispenzieri A, et al. High-dose melphalan and peripheral blood stem cell transplantation for light-chain amyloidosis with cardiac involvement. Blood. 2012;119(5):1117-22.
- Dispenzieri A, Gertz MA, Kyle RA, et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood. 2004;104:1881-7.
Summary
Amyloidosis refers to a rare group of disorders, which can involve various organs. It is caused by extracellular deposition of insoluble abnormal fibrils called amyloid. Cardiac amyloidosis in patients with AL-amyloidosis is common and associated with poor prognosis. Characteristic symptoms are low voltage ECG, echocardiographic findings of hypertrophy, diastolic dysfunction and symptoms of right heart failure. We present a 51 year-old patient with dyspnea and chest pain and recurring pleura effusions. ECG showed T-wave inversions in the lateral and inferior leads with normal QRS-amplitude and first echocardiography showed moderate concentric hypertrophy left ventricle, moderate enlarged atria and a restrictive diastolic filling pattern. The coronary angiography was normal. The diagnosis was delayed for several months and the symptoms worsened. Physicians must be aware of amyloidosis and other infiltrative cardiomyopathies as differential diagnosis in patients with unclear diastolic dysfunction even with normal QRS-amplitude.