





Mitokondriella sjukdomar kan orsaka svårt lidande och ibland tidig död. I dag saknas effektiva metoder för att behandla sjukdomarna.
Genom att byta ut mitokondrierna i samband med assisterad befruktning skulle man teoretiskt kunna förhindra överföring av mitokondriellt nedärvda sjukdomar från mor till barn.
Ännu har inget barn tillkommit genom tekniken, men de senaste åren har forskningsframsteg gjorts vid experiment på primater och humana celler. Det finns anledning att anta att kliniska försök snart inleds i Storbritannien.
Det finns kunskapsluckor om risker vid mitokondriebyte. Eftersom tekniken medför ärftliga genetiska förändringar, väcker den särskilda frågor av etisk karaktär.
Över hundra sjukdomar och syndrom finns under samlingsnamnet mitokondriella sjukdomar. Av dessa orsakas vissa av mutationer i cellkärnans DNA och andra av mutationer i mitokondriernas eget DNA (mtDNA). I båda fallen orsakar mutationerna defekter i energiomvandlingen (ATP-syntesen) i andningskedjan. De sjukdomar som orsakas av mutationer i mtDNA (mtDNA-sjukdom) nedärvs maternellt, eftersom endast äggets mitokondrier går över bestående till det blivande barnet vid befruktningen.
Vid mtDNA-sjukdom finns ofta en blandning av normalt och muterat mtDNA i olika proportioner i cellerna (s k heteroplasmi). Det förekommer även att allt mtDNA är muterat (s k homoplasmi). Nivån av heteroplasmi återspeglas till viss del i cellernas och vävnadernas funktion. Samma mutation kan yttra sig på flera olika sätt, vilket bl a beror på att nivån av heteroplasmi varierar hos olika individer. Symtomen kan dock vara olika även hos två individer med samma nivå av heteroplasmi, vilket medför att sjukdomens svårighetsgrad är svår att förutse genom genetisk diagnostik (t ex fosterdiagnostik).
Sjukdomsbilden vid mitokondriella sjukdomar är heterogen, men vissa vävnader och organ är särskilt drabbade eftersom de är extra känsliga för störningar i energiomsättningen, t ex centrala nervsystemet, muskler och njurar. Sjukdomen kan ibland visa sig redan vid födseln eller resultera i missfall under graviditeten. Hos en del visar sig symtomen senare i livet, ibland först vid vuxen ålder. Exempel på symtom vid mtDNA-sjukdom är påverkan på hörsel och syn, muskelförtvining, svaghet, hjärt-, lever- och njursjukdom, utvecklingsstörning, autism, epilepsi, diabetes och andra hormonella störningar.
Sjukdomarna kan vara lindriga men är ofta allvarliga, och många av de drabbade avlider i tidig ålder. I dag saknas effektiva metoder att behandla sjukdomarna.
En kvinna som har en viss nivå av heteroplasmi kan få ett barn som har högre eller lägre nivåer av muterat mtDNA än hon själv, och som därmed uppvisar en annan sjukdomsbild. Detta förklaras åtminstone delvis av en »flaskhalseffekt« som antas uppstå i den tidiga oogenesen och/eller embryogenesen, där endast ett fåtal slumpvist utvalda mitokondrier går vidare från modercellen till dottercellen [1, 2].
Den exakta prevalensen av mitokondriellt nedärvda sjukdomar är inte känd, men en uppskattning är ca 1 på 10 000 [3].
Situationen i dag för de patienter som vill ha barn
Barn som föds med allvarlig mitokondriell sjukdom riskerar svårt lidande och eventuellt tidig död. Gravida kvinnor som är bärare av mtDNA-sjukdom kan i vissa fall genomgå genetisk fosterdiagnostik i syfte att utröna det blivande barnets risk för att drabbas av sjukdom. Detta är endast möjligt om kvinnan bär på kända mutationer i mtDNA:t. Diagnostiken kan dock inte ge någon tillförlitlig sjukdomsprognos, eftersom det inte går att säkert förutsäga utifrån testresultatet hur det muterade mtDNA:t kommer att fördela sig i det blivande barnets kropp.
Fosterdiagnostik riskerar alltså att ställa föräldrarna inför ett svårt val mellan att avbryta eller fullfölja graviditeten, med en osäker riskbedömning som beslutsunderlag.
Preimplantatorisk genetisk diagnostik (PGD) har även prövats som verktyg för att minska risken för överföring av mitokondriellt nedärvd sjukdom [4, 5]. Vid PGD föreligger dock liknande problem som vid fosterdiagnostik, eftersom nivån av heteroplasmi i en enstaka cell från blastocysten kanske inte motsvarar nivån i det blivande barnets kropp; dessutom måste mutationen vara välkänd för att metoden ska vara möjlig. På grund av dessa problem erbjuds inte PGD till patienter med mtDNA-sjukdom i Sverige i dag.
Om en kvinna som bär på mtDNA-sjukdom vill ha barn i dag får hon och hennes partner alltså mer eller mindre chansa på att barnet inte kommer att drabbas av svår sjukdom. Om det genetiska bandet är av mindre betydelse för föräldrarna kan de använda sig av adoption eller äggdonation.
Mitokondriebyte för att förhindra överföring av sjukdom
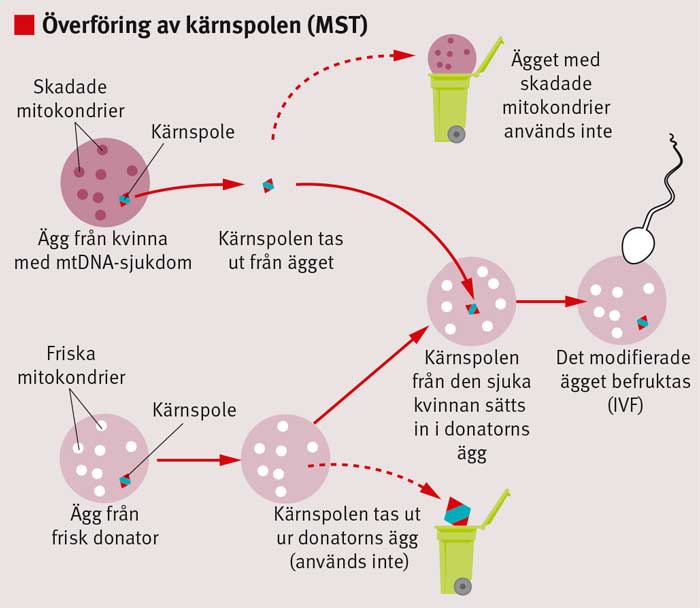
Genom s k mitokondriebyte skulle man eventuellt kunna förhindra att mtDNA-sjukdomar nedärvs från en kvinna till hennes barn. Principen innebär att man byter ut det skadade mtDNA:t från en drabbad kvinna till »friskt« mtDNA i samband med in vitro-fertilisering (IVF). Mitokondriebytet utförs genom modifiering av antingen obefruktade eller befruktade ägg (Figur 1 och 2).
Vid modifiering av obefruktade ägg är överföring av kärnspolen (maternal spindle transfer, MST) den metod som för närvarande verkar mest lovande. Vid MST överförs kärnspolen, till vilken kromosomerna är bundna, i ett ägg från en drabbad kvinna till ett donerat ägg som i sin tur tömts på sin kärnspole (Figur 1). Det donerade ägget kommer att innehålla kromosomer från den drabbade kvinnan och »friska« mitokondrier från äggdonatorn, och det kan befruktas med spermier från den tilltänkte fadern.
Vid modifiering av befruktade ägg överförs prokärnorna i ett befruktat ägg från ett par där kvinnan bär på mtDNA-sjukdom till ett befruktat ägg som bildats utifrån ett donerat ägg (pronuclear transfer, PNT) (Figur 2). Med denna metod erhålls samma resultat som vid MST.
MST och PNT har i forskningsstudier utförts på djur (exempelvis möss och makaker) och på obefruktade och befruktade ägg från människor. Forskningen på området är ännu begränsad, och inget människobarn har blivit till med hjälp av teknikerna hittills. Dock rapporterades det i medier år 2003 att ett forskarteam i Kina låtit återföra fem befruktade ägg som varit föremål för MST till en kvinnas livmoder, vilket resulterade i en trillinggraviditet [6]. Enligt medierapporteringen resulterade graviditeten i missfall efter några månader.
Metodernas effektivitet för att byta ut mitokondrierna
Forskningsstudier har visat på ett problem hos tekniker för mitokondriebyte, vilket innebär att mitokondrierna inte alltid byts ut till 100 procent. Ofta medföljer en liten mängd cytoplasma innehållande de ursprungliga mitokondrierna vid både MST och PNT. Omfattningen av denna oavsiktliga överföring är av stor betydelse, eftersom den är relaterad till risken för sjukdom hos den blivande individen.
PNT. Tekniken PNT har använts på möss sedan 1980-talet, och flera experiment har visat att metoden fungerar för att byta ut mtDNA i befruktade ägg [1, 7]. Storleken på överföringen av oönskat mtDNA från den ursprungliga äggcellen har varierat i dessa studier. PNT har också utförts på humana befruktade ägg, vilket resulterade i normal blastocystutveckling upp till 6–8 dagar efter överföringen [8]. Blastocysterna i denna studie innehöll i genomsnitt <2 procent av oavsiktligt överfört mtDNA.
MST. År 2009 utfördes MST på primater (rhesusmakaker) av en amerikansk forskargrupp [9]. De befruktade äggen utvecklades normalt och användes till att inducera dräktighet, vilket resulterade i fyra friska avkommor. Ingen överföring av oönskat mtDNA kunde observeras hos någon av ungarna (dock användes en teknik som endast kunde påvisa ned till 3 procent av det totala mtDNA:t). Makakungarnas utveckling har följts upp till 3 års ålder, och hittills har inga avvikelser rapporterats jämfört med kontroller [10]; inget oönskat mtDNA har heller kunnat påvisas. I en annan studie utvärderades överföring av oönskat mtDNA i många olika organ hos makakfoster som utvecklats efter MST [2]. I alla organ kunde endast låga (<0,5 procent) eller icke-detekterbara nivåer av oönskat mtDNA påvisas, förutom i de fetala oocyterna där nivåer kring ca 15 procent uppmättes i vissa av proven.
Samma forskargrupp har även utfört MST på humana äggceller som sedan befruktades, vilket resulterade i normal utveckling till blastocyster med i genomsnitt 0,5 procent oavsiktligt överfört mtDNA [10].
En annan forskargrupp har utfört en variant av MST, där man tillämpade frysning eller nedkylning av humana äggceller för att depolymerisera kärnspolen före överföringen [11]. Efter artificiell aktivering (äggen befruktades inte i denna studie) sågs normal utveckling till blastocyster med <1 procent oavsiktligt överfört mtDNA. Från blastocysterna genererades stamcellslinjer, som också innehöll låga nivåer av oönskat mtDNA, och nivåerna sjönk till icke-detekterbara under odling. Nivåerna låg kvar under detektionsgränsen efter >1 års odling av stamcellerna och även efter differentiering till olika celltyper.
Risker förknippade med överföring av oönskat mtDNA
De ovan nämnda studierna tyder på att en tämligen låg grad av överföring av oönskat mtDNA kan förväntas vid optimering av teknikerna MST och PNT (ca 0–0,5 procent respektive ca 2 procent). Sannolikheten för att sjukdom ska inträffa vid så låga proportioner av muterat mtDNA är troligtvis liten. Exempelvis uppskattades i en studie som avsåg att vara vägledande inför PGD sannolikheten för att inte drabbas av sjukdom till 95 procent om nivån av heteroplasmi i cellerna var <18 procent [5].
Studierna på makaker och stamcellslinjer tyder på att det inte sker någon betydande ackumulering av muterat mtDNA i cellerna om det har överförts i låga nivåer [10, 11]. Däremot kan den s k flaskhalseffekten vid oogenesen medföra att vissa äggceller får märkbart högre proportioner av muterat mtDNA än de somatiska cellerna, vilket även observerades i studien av fetal makakvävnad [2]. Det betyder att det, även om man genom MST eller PNT skulle lyckas med att förhindra sjukdomsuppkomst hos ett barn, fortfarande kan finnas risk för att sjukdom uppstår hos kommande generationer om barnet är en flicka.
I en studie där man undersökte ärftlighetsmönster för en mtDNA-sjukdom med en klinisk tröskel på ca 60 procent (stor risk för sjukdom vid >60 procent muterat mtDNA) uppskattades risken för sjukdom i nästkommande generationer dock vara mycket låg vid överföring av <3 procent oönskat DNA [12].
Om risken för sjukdomsuppkomst i påföljande generationer vill undvikas helt, skulle man genom könsselektion kunna välja ut befruktade ägg med Y-kromosom för återföring till livmodern. En sådan könsselektion kan anses som etiskt kontroversiell, men bör sättas i relation till att det är tillåtet att använda könsselektion för att undvika allvarliga könsbundna sjukdomar.
Effekter av att introducera främmande mtDNA i cellen
I en forskningsstudie observerades fysiologiska nedsättningar hos möss som med hjälp av PNT hade producerats för att innehålla två olika sorters mtDNA [13]. I det aktuella experimentet användes mtDNA i proportioner om ca 50–50 procent från två mustyper som genetiskt sett var mycket olika varandra, men båda typerna av mtDNA var normala (ej sjukdomsalstrande) var och en för sig. Resultaten tolkades i studien som att det kan vara olyckligt att skapa heteroplasmi av två sorters mtDNA som skiljer sig mycket åt. Dock visar forskning att MST och PNT kan generera mycket lägre nivåer av heteroplasmi än 50 procent om tekniken optimeras. Om man vill undvika att skapa heteroplasmi av två sorters mtDNA med stor genetisk skillnad vid eventuell användning av tekniken på människor, skulle man kunna välja att använda donerat mtDNA som liknar den sjukdomsdrabbade individens.
Farhågor har också uttryckts om att interaktionen mellan kärn-DNA och mtDNA skulle kunna påverkas negativt av att introducera »främmande« mtDNA i cellen. När makak-ungarna genererades användes dock två sorters mtDNA med hög genetisk diversitet (den genetiska skillnaden uppges vara större än de skillnader som kan finnas i mtDNA mellan olika människor), och inga problem har observerats hos ungarna (som dock inte var fler än fyra) upp till 3 års ålder [10]. I denna diskussion brukar det även påpekas att vid en vanlig befruktning är hälften av kärn-DNA:t – det som kommer från fadern – i princip främmande för mtDNA:t, vilket inte orsakar några (kända) problem.
Övriga risker
Kromosomskador skulle teoretiskt sett kunna induceras av de reagenser som används vid MST och PNT, eller av någon annan komponent i proceduren. Även om ingen förhöjd risk för kromosomskador observerades hos makak-ungarna [9] eller i de humana stamcellslinjer som tillkommit efter MST [11], är forskningsunderlaget mycket begränsat.
En annan farhåga som har uttryckts är om mikromanipuleringen av cellerna vid PNT och MST skulle kunna inducera epigenetiska förändringar, eftersom ingreppet sker i ett känsligt skede under äggets epigenetiska omprogrammering. Konventionell IVF-behandling har associerats med viss ökad risk för sällsynta epigenetiska sjukdomar, även om den totala risken för sjukdomarna fortfarande är mycket låg [14, 15]. I de ovan nämnda studierna av MST och PNT har man inte specifikt letat efter epigenetiska förändringar, men de makaker som har tillkommit med metoden har bedömts vara friska, och de stamcellslinjer som genererats har haft ett till synes normalt genuttryck [10, 11]. För att kunna upptäcka eventuella epigenetiska förändringar till följd av MST och PNT skulle det behövas långtidsuppföljningar av ett stort antal individer som tillkommit med tekniken.
Utvecklingen i Storbritannien
I dag är tekniker för mitokondriebyte förbjudna att använda i Sverige enligt lagen om genetisk integritet (2006:351), eftersom metoden medför genetiska förändringar som kan gå i arv. För att tekniken skulle kunna användas inom forskning eller behandling skulle alltså en lagändring krävas.
I Storbritannien har Human Reproduction and Fertilisation Authority (HFEA) på uppdrag av den brittiska regeringen utvärderat aktuell forskning om MST och PNT med fokus på eventuella medicinska risker [16, 17]. I rekommendationen till regeringen konstaterade man bl a att forskningsresultaten hittills inte tyder på att MST eller PNT skulle vara osäkra metoder. Man specificerade dock ett antal ytterligare forskningsprojekt som rekommenderas att utföra innan metoden prövas kliniskt på människor.
HFEA fick även i uppdrag att genomföra ett omfattande offentligt samråd (public consultation) om frågan, och från detta drog man slutsatsen att det fanns ett gott stöd för tekniken hos allmänheten [18].
Det brittiska etikrådet Nuffield Council on Bioethics har också analyserat frågan ingående och kommit fram till att tekniken är godtagbar om den kan bedömas som effektiv och tillräckligt säker [19].
Storbritanniens regering meddelade i juli 2013 att man avser att ta fram ett lagförslag som tillåter mitokondriebyte under strikta former [20]. Förslaget förväntas behandlas i parlamentet under 2014. Det finns alltså anledning att anta att kliniska försök med mitokondriebyte kan komma att påbörjas i Storbritannien inom en relativt nära framtid. Utvecklingen i Storbritannien har fått stor internationell uppmärksamhet och väckt debatt både angående den medicinska bedömningen [21] och de mer principiella frågorna kring tekniken [22].
Även i USA är tekniken mitokondriebyte under diskussion, och frågan behandlades vid ett möte hos läkemedelsmyndigheten Food and Drug Administration (FDA) tidigare i år [23].
Etiska aspekter på mitokondriebyte
Mitokondriebyte aktualiserar den etiska konflikten mellan intresset att undvika sjukdom och lidande hos ett blivande barn och risken för att utsätta barnet för okända negativa effekter som tekniken eventuellt kan medföra. Att de genetiska förändringar som introduceras vid mitokondriebyte kan gå i arv innebär att sjukdom skulle kunna undvikas även hos kommande generationer, men det betyder också att eventuella risker som är förknippade med metoden kan föras vidare.
Liksom i många andra situationer innebär det vissa risker att gå från experimentell forskning på celler och djur till kliniska försök på människor. Att avgöra när det är dags att ta steget blir i dessa fall en bedömningsfråga utifrån tillgängligt forskningsunderlag och utifrån övriga risker och möjligheter som är förknippade med att använda respektive avstå från metoden.
Den sammanvägda riskbild som finns vid mitokondriebyte – ofullständigt byte av mitokondrierna, kromosomavvikelser, eventuella störningar i interaktionen mellan kärn-DNA och mtDNA, epigenetiska störningar och okända risker – ska vägas mot de mer kända risker som en mitokondriell sjukdom innebär, dvs svårt lidande och tidig död i de allvarligaste fallen. En kärnfråga i detta sammanhang är vilka förväntade biverkningsrisker som skulle kunna vara förenliga med en ansvarsfull klinisk användning av tekniken. Kunskapsunderlaget är ännu alltför bristfälligt för att kunna göra en sådan avvägning.
Ett mer principiellt etiskt problem är om det över huvud taget är acceptabelt att genetiskt modifiera befruktade eller obefruktade ägg i syfte att undvika sjukdom. Det kan argumenteras att en gräns mot genetisk modifiering av människor passeras om mitokondriebyte tillåts, vilket av olika skäl kan anses oacceptabelt. Argumentet kan bottna i en rädsla för att varje form av genmodifiering på sikt skulle kunna leda till en glidning mot ökad acceptans avseende modifiering och förädling av människan, ofta kallad »det sluttande planet«. Det sluttande planet-argumentet skulle även kunna beskrivas som en typ av riskvärdering med avseende på risken att en metod öppnar upp för andra metoder och tillämpningar.
När det gäller mitokondriebyte är metoden dock tydligt avgränsad till att beröra enbart mitokondriellt DNA, och metoden skulle inte kunna användas för att t ex modifiera gener i cellkärnan. Denna distinkta tekniska avgränsning skulle kunna innebära att en skiljelinje mot en vidare användning av genmodifiering med större sannolikhet kan upprätthållas.
En annan aspekt som har diskuterats mycket internationellt är huruvida de barn som tillkommer genom mitokondriebyte skulle få »tre genetiska föräldrar« och vilka etiska problem det skulle kunna innebära [24]. Den genetiska komponent som finns i mitokondrien är dock mycket liten i förhållande till cellens totala DNA – 37 gener jämfört med ca 20 000 transkriberade gener i cellkärnan [25]. Såvitt man vet påverkar inte mtDNA andra funktioner än de som hör till mitokondrien.
Smer avråder – osäkerhet om de medicinska riskerna
När Statens medicinsk-etiska råd (Smer) analyserade frågan [26] kom rådet fram till att mitokondriebyte inte var etiskt godtagbart mot bakgrund av den kunskapsosäkerhet som i dag råder om de medicinska riskerna. I bedömningen vägde rådet in att risken för allvarlig sjukdom visserligen kan vara stor för de barn som föds med muterat mtDNA, men också att det finns alternativa möjligheter för drabbade föräldrar att med hjälp av äggdonation eller adoption få barn utan mitokondriella sjukdomar. Rådet ansåg vidare att de barn som eventuellt tillkommer genom mitokondriebyte bör anses ha två genetiska föräldrar och inte tre.
Rådet var dock inte enigt angående den principiella frågan. Här ansåg en majoritet av rådets ledamöter att tekniken skulle kunna vara godtagbar under förutsättning att metoden utvecklas så att riskerna bedöms som små. Huvudargumenten var att mycket svåra sjukdomar skulle kunna undvikas – sannolikt också i kommande generationer – och att byte av mtDNA mycket tydligt kan avgränsas mot ingrepp i kärn-DNA. En minoritet av ledamöterna tog principiellt avstånd från tekniken. De ansåg att mitokondriebyte på sikt skulle kunna innebära ett hot mot människovärdet och att sluttande planet-argumentet är högst relevant.
I rapporten efterlyser Smer en samhällelig diskussion om genterapi för att undvika allvarliga sjukdomar, ett ämne som inte har debatterats nämnvärt de senaste åren. Att debatten är så ljum skulle möjligen kunna avspegla en allmänt mindre ifrågasättande inställning till medicinska frontlinjeterapier såsom somatisk genterapi, som också kan anses beröra etiska värden.
Om nu de alltför stora kunskapsluckorna varit avgörande för Smer:s ställningstagande, hur ska kunskapsluckorna då täppas igen? Den brittiska rapporten om mitokondriebyte [17] lyfter fram framför allt behovet av forskning på befruktade och obefruktade humana ägg och stamceller från humana ägg samt experiment i primatmodeller. Att Smer avråder från mitokondriebyte i svensk klinisk praxis i dag innebär inte på något sätt ett ställningstagande mot dessa typer av forskning.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Referenser
- Jenuth JP, Peterson AC, Fu K, et al. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet. 1996;14(2):146-51.
- Lee HS, Ma H, Juanes RC, Tachibana M, et al. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell Rep. 2012;1(5):506-15.
- Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63(1):35-9.
- Bredenoord AL, Dondorp W, Pennings G, et al. PGD to reduce reproductive risk: the case of mitochondrial DNA disorders. Hum Reprod. 2008;23(11):2392-401.
- Hellebrekers DM, Wolfe R, Hendrickx AT, et al. PGD and heteroplasmic mitochondrial DNA point mutations: a systematic review estimating the chance of healthy offspring. Hum Reprod Update. 2012;18(4):341-9.
- BBC News. Foetus with three parents created. 10 okt 2003 [citerat 4 dec 2013]. http://news.bbc.co.uk/2/hi/health/3189718.stm
- Sato A, Kono T, Nakada K, et al. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci U S A. 2005;102(46):16765-70.
- Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465(7294):82-5.
- Tachibana M, Sparman M, Sritanaudomchai H, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461(7262):367-72.
- Tachibana M, Amato P, Sparman M, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493(7434):627-31.
- Paull D, Emmanuele V, Weiss KA, et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493(7434):632-7.
- Samuels DC, Wonnapinij P, Chinnery PF. Preventing the transmission of pathogenic mitochondrial DNA mutations: can we achieve long-term benefits from germ-line gene transfer? Hum Reprod. 2013;28(3):554-9.
- Sharpley MS, Marciniak C, Eckel-Mahan K, et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151(2):333-43.
- Källén B, Finnström O, Lindam A, et al. Congenital malformations in infants born after in vitro fertilization in Sweden. Birth Defects Res A Clin Mol Teratol. 2010;88(3):137-43.
- van Montfoort AP, Hanssen LL, de Sutter P, et al. Assisted reproduction treatment and epigenetic inheritance. Hum Reprod Update. 2012;18(2):171-97.
- Haites N, Lovell-Badge R. Scientific review of the safety and efficacy of methods to avoid mitochondrial disease through assisted conception. London: Human Fertilisation and Embryology Authority; 2011.
- Haites N. Scientific review of the safety and efficacy of methods to avoid mitochondrial disease through assisted conception: update. London: Human Fertilisation and Embryology Authority; 2013.
- Human Fertilisation and Embryology Authority. Mitochondria replacement consultation: Advice to Government. Mars 2013 [citerat 4 dec 2013].
- Novel techniques for the prevention of mitochondrial DNA disorders: an ethical review. London: Nuffield Council on Bioethics; 2012.
- UK Department of Health/Human Fertilisation and Embryology Authority. Innovative genetic treatment to prevent mitochondrial disease [pressmeddelande]. 28 jun 2013 [citerat 4 dec 2013]. https://www.gov.uk/government/news/innovative-genetic-treatment-to-prevent-mitochondrial-disease
- Reinhardt K, Dowling DK, Morrow EH. Mitochondrial replacement, evolution, and the clinic. Science. 2013;341 (6152):1345-6.
- Darnovsky M. A slippery slope to human germline modification. Nature. 2013;499(7457):127.
- US Food and Drug Administration. Advisory Committee Calendar. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm380042.htm
- Baylis F. The ethics of creating children with three genetic parents. Reprod Biomed Online. 2013;26(6):531-4.
- McHale CM, Zhang L, Thomas R, et al. Analysis of the transcriptome in molecular epidemiology studies. Environ Mol Mutagen. 2013;54(7):500-17.
- Smer. Mitokondriebyte för att undvika allvarlig ärftlig sjukdom – etiska aspekter. Stockholm: Statens medicinsk-etiska råd; 2013. Rapport 2013:2.
Summary
Mitochondrial inherited disorders may cause severe disease, suffering and premature death. Today, there are no effective treatments available for the conditions. Novel techniques involving replacement of mutated mitochondria with mitochondria from healthy donors in association with IVF may provide a possibility to prevent children from inheriting the disorders. Recent research results from studies in primates and human cells have been reasonably promising. However, no baby has yet been born through the technique, and possible short and long term risks in humans are difficult to predict. Furthermore, the technique causes genetic alterations that are inherited, and thus raises fundamental ethical questions. The Swedish National Council on Medical Ethics has advised that mitochondrial replacement should not be used in humans in Sweden until more scientific knowledge is available.


















