
Från 1 januari 2019 har en ny särskild ICD-10-kod (E78.0A) införts för den ärftliga blodfettrubbningen familjär hyperkolesterolemi. Patienter med sjukdomen har kraftigt ökad risk att tidigt, ofta före 50-årsåldern, utveckla allvarlig aterosklerotisk sjukdom såsom hjärtinfarkt. Detta kan förhindras genom tidigt insatt behandling.
Socialstyrelsen gav i sina riktlinjer från 2015 hög prioritet till vården att identifiera och diagnostisera personer och släkter med sjukdomen.
Koden E78.0A kan, vid rätt användning, bli ett effektivt verktyg för det arbetet, bl a genom att underlätta säkerställandet av kaskadscreening och uppfyllelse av behandlingsmål.
Familjär hyperkolesterolemi är en viktig riskfaktor för kardiovaskulär sjukdom tillsammans med rökning, hypertoni och diabetes [1, 2]. Familjär hyperkolesterolemi är en autosomalt dominant nedärvd blodfettsrubbning som finns i en extremt ovanlig homozygot form (ca 1 på miljonen) och i en vanligare heterozygot form (ca 1/250–400).
Sjukdomen kan i de flesta fall förklaras av en mutation i någon av de tre generna som kodar för LDL-receptorn (LDLR), apolipoprotein B (APOB) eller proteinet PCSK-9 (PCSK9), vilka alla påverkar leverns upptag och reglering av kolesterol i blodet. En patient med heterozygot familjär hyperkolesterolemi uppvisar ofta dubblerad plasmanivå av kolesterol jämfört med en frisk person, men variationen av nivåer är stor inom populationen av patienter med familjär hyperkolesterolemi. I Tabell 1 finns både referensvärden för LDL-kolesterol i olika åldrar samt nivån av LDL-kolesterol hos obehandlade patienter med genetiskt verifierad heterozygot familjär hyperkolesterolemi [3].
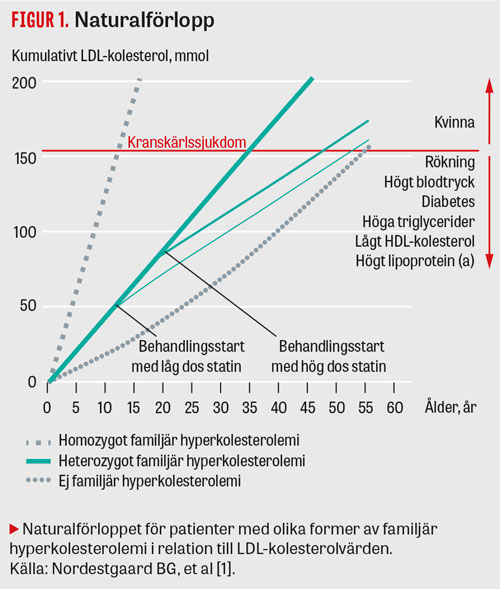
Sjukdomen medför uppskattningsvis 5–10 gånger ökad risk att utveckla en allvarlig kardiovaskulär åkomma jämfört med normalbefolkningen. Betydelsen av livstidsexponeringen av ett förhöjt LDL-kolesterolvärde för aterosklerosutveckling hos patienter med familjär hyperkolesterolemi jämfört med en frisk individ illustreras i Figur 1. Samverkan mellan kolesterolnivån och de övriga klassiska riskfaktorerna påverkar tillsammans med tidpunkten för insatt medicinsk behandling progressen av sjukdomen.
Den ökade hjärt–kärlrisken understryks ytterligare i en registerstudie från Norge där författarna kunde visa att 93 procent av patienterna med familjär hyperkolesterolemi hade en hjärt–kärlsjukdomsdiagnos vid sin död och att medelåldern för första händelsen var 44 år [4], även om familjär hyperkolesterolemi inte verkar öka risken för ischemisk stroke hos patienter utan annan aterosklerotisk manifestation [5].
Konsekvenserna av familjär hyperkolesterolemi är behandlingsbara, och tidigt insatt behandling påverkar kraftigt positivt sjukdomens naturalförlopp. Detta är en av anledningarna till att Socialstyrelsen i sina nationella riktlinjer från 2015 gav hög prioritet åt arbetet med att diagnostisera patienter med sjukdomen och identifiera sjuka släktingar [6].
Diagnostiken grundar sig på noggrann sammanvägning
Diagnostiken av familjär hyperkolesterolemi grundar sig främst på en noggrann sammanvägning av patientens kardiovaskulära hereditet, anamnes, status och blodfettsvärden. Det är i huvudsak lipoproteinet LDL (low density lipoproteins) som ansvarar för kolesteroltransporten i plasma, och LDL-kolesterolnivåerna är ofta kraftigt förhöjda hos patienter med familjär hyperkolesterolemi.
Det är däremot en spridd missuppfattning att diagnosen kan baseras på endast ett enstaka högt LDL-kolesterolvärde. Anledningen till detta är att patientens hela blodfettsprofil inklusive triglyceridvärde bör kontrolleras åtminstone två gånger för att utesluta mätfel och naturlig biologisk variation. Dessutom är överlappningen stor i referensvärden för LDL-kolesterol för friska och sjuka (Tabell 1), vilket gör det svårt att diskriminera mellan de båda grupperna.
Hereditetens betydelse vid anamnesupptagning kan således inte nog poängteras. Familjär hyperkolesterolemi karaktäriseras av ett dominant ärftlighetsmönster med hög penetrans, vilket gör att man i princip alltid kan utgå från att sjukdomen finns hos en av föräldrarna om man diagnostiserar den hos sin patient. Vi har i Fakta 1 sammanställt en lista över fynd hos patienten som, när de förekommer tillsammans, starkt talar för diagnosen.
Det finns flera sätt att förbättra precisionen i diagnostiken. Ett praktiskt och validerat kliniskt hjälpmedel är skattningsinstrumentet från Dutch Lipid Clinic Network (DLCN), som finns tillgängligt bl a som ett webbverktyg [7] som är rekommenderat av European Atherosclerosis Society (EAS). Instrumentet är i praktiken ett poängsättningssystem för sammanvägning av patientens uppgifter baserat på anamnes inklusive hereditet, kliniskt status och blodprov. DLCN-poäng >8 anses liktydigt med diagnosen, men många, särskilt yngre patienter (<40 år), har lägre poäng.
Någon entydig beslutsgräns för att ställa diagnosen finns således inte. En av instrumentets största svagheter är att det underskattar yngre personers sannolikhet för familjär hyperkolesterolemi, och det kan inte heller användas på barn. Vår uppfattning är dock att alla vuxna patienter där man överväger diagnosen ska graderas enligt DLCN-skalan, eftersom den ökar objektiviteten i bedömningen.
Genetisk utredning är ett viktigt tillskott
Genetisk utredning är ett viktigt tillskott till diagnostiken. Olika metoder har funnits under lång tid men har först nyligen börjat användas i större omfattning i klinisk praxis. I dagsläget saknas nationella riktlinjer för när och hur genetisk testning ska användas i diagnostiken. Tillgången till testning är högst varierande, och kostnaderna skiljer sig också kraftigt mellan olika metoder, där en enklare mutationskonfirmation kan kosta ca 1 500 kronor, medan djupsekvensering av alla kända gener för familjär hyperkolesterolemi på vissa laboratorier kan kosta 20 000–30 000 kronor.
Vi rekommenderar att bara vissa enheter i varje region med kunskap om den genetiska utredningens styrkor och begränsningar ska remittera patienter för genetisk utredning. I Fakta 2 finns en sammanställning på möjliga indikationer för genetisk testning och där nyttan av testningen är som störst.
En viktig kunskap om genetisk utredning är att metoden inte kan utesluta familjär hyperkolesterolemi utan endast bekräfta den. Därför är fynd av en patologisk mutation inte ett krav för att kunna ställa diagnosen. Samtidigt är den största risken med genetisk utredning att patienter med »äkta« familjär hyperkolesterolemi felaktigt kan frikännas från diagnosen baserat på ett negativt gentest och felaktig beslutslogik hos den oinitierade läkaren.
Fördelarna med en genverifierad diagnos är att den kan uppfattas som »mer säker« av både behandlande läkare och patient, vilket underlättar screening av indexpatientens släktingar och följsamheten till den livslånga medicinska behandlingen.
Behandlingen kräver specialistresurser
Den homozygota formen av familjär hyperkolesterolemi kräver specialistklinikens alla resurser, där farmakologisk behandling ofta måste kombineras med lipoproteinaferes 1–2 gånger/vecka (en metod att rena blodet från LDL-partiklar) för att hålla kolesterolnivåerna på en acceptabel nivå.
Den vanligare heterozygota formen svarar ofta bra på en kombinationsbehandling av en hög dos av en effektiv statin och en kolesterolupptagshämmare (ezetimib). Primärpreventiv behandling är i princip alltid indicerad, och behandlingsmål för LDL-kolesterol är enligt fastställda europeiska riktlinjer ≤2,5 mmol/l för vuxna och ≤3,5 mmol/l för barn.
Långtidsuppföljningsstudier (>20 år) av barn visar att tidigt insatt läkemedelsbehandling är av värde och således bör övervägas redan från (8–)10 års ålder. Likaså kan aterosklerotiska kärlförändringar ses i karotisartären redan hos barn i 8–10-årsåldern. Behandling av barn ska åtminstone initialt skötas av barnläkare.
För vuxna patienter som inte når behandlingsmålet för LDL-kolesterol med konventionell behandling (statin och ezetimib) finns möjlighet att förstärka den primärpreventiva behandlingen med en PCSK9-hämmare om LDL-kolesterol är ≥3 mmol/l. De flesta av dessa fall handlar om patienter som är totalt statinintoleranta och har kvarvarande höga LDL-kolesterolvärden (>5–6 mmol/l). PCSK9-hämmarna halverar plasmakolesterolnivåerna genom uppreglering av antalet kolesterolreceptorer i levern. Behandlingen är dyr och ska initieras av en specialist i endokrinologi, kardiologi eller internmedicin.
Kvinnor i fertil ålder ska informeras om att statiner, ezetimib och PCSK9-hämmare är kontraindicerade vid graviditet och amning. Uppehåll får göras under graviditet och amning med tydlig instruktion om att återuppta behandling i nära anslutning till avslutad och gärna förkortad amning (≤6 månader).
Kaskadscreening bör helst samordnas av koordinator
När en patient erhåller diagnosen familjär hyperkolesterolemi får det konsekvenser inte bara för patienten utan också för dennes släkt. Sjukdomens autosomalt dominanta nedärvningsmönster innebär att sannolikheten är 50 procent för att förstagradssläktingar bär på samma patologiska mutation.
I praktiken blir det indexpatientens uppgift att kontakta sina släktingar och informera om sjukdomen, eftersom den läkare som fastställt diagnosen inte är behandlingsansvarig för släktingar och inte heller har rätt att kontakta dem. Exakt hur denna kaskadscreeningsprocess sköts varierar över landet. I den ideala situationen finns en koordinator som kan underlätta processen för att uppnå god täckningsgrad och kostnadseffektivitet.
A:et spelar roll
I dag används den generiska ICD-10-koden E78.0 för alla former av hyperkolesterolemi. I praktiken används koden sannolikt ofta godtyckligt för att avspegla både sekundära former av hyperkolesterolemi (sekundära till t ex kost eller sjukdom) eller för att markera att patienten har ett kolesterolvärde ovanför önskad nivå sett ur ett sekundärpreventivt perspektiv. Värdet av att tilldela en patient diagnosen är i praktiken ringa, eftersom den inte säger någonting om orsaken till kolesterolstegringen eller ens om det finns en kolesterolstegring över huvud taget och om medicinsk behandling ska ges.
Diagnosen familjär hyperkolesterolemi tilldelas från årsskiftet 2018–2019 en specifik och unik ICD-10-kod: E78.0A. Till skillnad från den generiska koden E78.0 kan den nya koden ha ett starkt signalvärde för både läkare och patient och vara ett verktyg för förbättrad vård. För den behandlande klinikern innebär beslutet att ställa diagnosen familjär hyperkolesterolemi att primärpreventiv kolesterolsänkande behandling behöver initieras enligt evidensbaserade riktlinjer. Hos patienten höjs medvetenheten om behovet av livslång och intensiv farmakologisk behandling.
En specifik diagnos blir också ett startskott för kaskadscreening för att identifiera eventuella sjuka släktingar och underlättar spridandet av budskapet om en kronisk men behandlingsbar sjukdom.
E78.0A – endast för familjär hyperkolesterolemi
I framtiden kommer möjligheter att utvärdera den svenska vården av patienter med familjär hyperkolesterolemi att bli tillgängliga genom sammanställningar av data från nationella kvalitetsregister och Socialstyrelsens diagnosregister. Den specifika diagnosen blir då ett kraftfullt redskap för att säkerställa att vården är god, jämlik över landet och förbättras över tid.
Det är därför vår starka uppfattning att diagnoskoden E78.0A ska användas endast då diagnosen familjär hyperkolesterolemi är säkerställd, antingen via klinisk bedömning eller via genetisk verifiering av en patologisk mutation. Patientens vårdgivare (primärvårds- eller sjukhusbaserad) bör remittera patienten till en klinik med lipidspecialisering (endokrinologi, internmedicin eller kardiologi) för hjälp med diagnostik och behandling.
Den specifika diagnoskoden bör inte användas för patienter med höga blodfetter som inte uppfyller övriga kriterier för den formella diagnosen familjär hyperkolesterolemi. Detta skulle riskera att minska användbarheten för diagnosen och respekten för de »förpliktelser« som föreligger hos både patient och behandlande läkare, vilka syftar till framgångsrik släktscreening och livslång och effektiv medicinsk behandling.
Potentiella bindningar eller jävsförhållanden: Jonas Brinck och Mats Eriksson har akademiska forskningsanslag från Amgen och Sanofi.
Fakta 1.
Fynd som sammantagna talar starkt för diagnosen familjär hyperkolesterolemi
- P-LDL-kolesterol ≥5,0 mmol/l hos patient utan lipidsänkande behandling
- Hos patienten förekomst av tidig1 aterosklerotisk hjärt- och kärlsjukdom eller hereditet för tidig1 kranskärlssjukdom eller hjärt–kärldöd hos förstagradssläkting
- Avsaknad av förhöjda P-triglyceridvärden
1 Tidig definieras som händelse före 55 års ålder hos män och före 60 års ålder hos kvinnor
Fakta 2.
Faktorer som stärker indikationen för genetisk testning av familjär hyperkolesterolemi
- Förstärkning av klinisk diagnostik av familjär hyperkolesterolemi för patienter med DLCN 5–7 poäng (4 p om <40 år)
- Fastställande av diagnos hos indexpatient (dvs den första identifierade patienten i en nyupptäckt släkt med familjär hyperkolesterolemi)
- Konfirmation av mutation hos släktingar med känd diagnos familjär hyperkolesterolemi, speciellt hos barn och yngre (<20 år)
Referenser
- Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478-90a.
- Yusuf S, Hawken S, Ounpuu S, et al; INTERHEART Study Investigators. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet. 2005;366(9497):1640-9.
- Huijgen R, Kindt I, Verhoeven SB, et al. Two years after molecular diagnosis of familial hypercholesterolemia: majority on cholesterol-lowering treatment but a minority reaches treatment goal. PLoS One. 2010;5(2):e9220.
- Krogh HW, Mundal L, Holven KB, et al. Patients with familial hypercholesterolaemia are characterized by presence of cardiovascular disease at the time of death. Eur Heart J. 2016;37(17):1398-405.
- Beheshti S, Madsen CM, Varbo A, et al. Relationship of familial hypercholesterolemia and high low-density lipoprotein cholesterol to ischemic stroke. Circulation. 2018;138(6):578-89.
- Socialstyrelsen. Nationella riktlinjer. Familjär hyperkolesterolemi, familjära kardiomyopatier och jonkanalsjukdomar samt familjär aortasjukdom. https://www.socialstyrelsen.se/nationellariktlinjerhjartsjukvard/sokiriktlinjerna/familjarhyperkolesterolemi-fam
- FH score. https://www.fhscore.eu/#/fhQuestionnaire
Summary
At the turn of the year 2018/19, a new ICD-10 code (E78.0A) will be introduced in Sweden for the hereditary blood lipid disorder familial hypercholesterolemia (FH). Patients with FH have a significantly increased risk of developing atherosclerotic disease, such as myocardial infarction before the age of 50. However, early diagnosis and start of treatment of FH can ameliorate the disease’s negative long term effects. The Swedish National Board of Health and Welfare gave in its guidelines from 2015 a high priority to the work of identifying and diagnosing individuals with FH in the general population. The introduction of the ICD-10 code E78.0A for FH may, when properly used, be an effective tool in this work.
Sedan åtskilliga år har jag tillsammans med tre internationella experter granskat litteraturen kring familjär hyperkolesterolämi (FH) i detalj.1 Vad vi har funnit är att hjärtdödligheten hos individer med denna abnormitet inte beror på högt kolesterol utan på förhöjda koagulationsvärden. Vi har till exempel funnit flera kohortstudier som visat att LDL-kolesterolet hos FH individer med hjärtinfarkt inte är högre än hos FH individer i samma åldersklass utan hjärtinfarkt. Åtskilliga studier har dessutom visat att FH individer med de högsta kolesterolvärdena inte är mer åderförkalkade än FH individer med de lägsta värden. Det är endast en liten minoritet som dör tidigt av hjärtinfarkt. I snitt lever människor med FH lika länge som andra. Några få dör tidigt av hjärtinfarkt medan äldre med FH lever längre än normalt därför att deras höga kolesterol skyddar mot cancer och infektionssjukdomar. Det finns tio kontrollerade och randomiserade kolesterolsänkande experiment. Ingen av dem har lyckats minska risken för kardiovaskulär sjukdom. Den enda behandlingen som lyckats är aferes, troligen därför att man även avlägsnar många koagulationsfaktorer med denna teknik.
De abnorma koagulationsfaktorerna inkluderar bland annat högt fibrinogen, hög faktor VIII, högt protrombin och olika trombocytabnormaliteter. Tydligen är det endast några få som ärver detta.
Våra fynd överensstämmer med det jag tillsammans med 16 internationella experter har dokumenterat; nämligen att kolesterolhypotesen inte tillfredsställer en enda av Bradford Hill´s kriterier för en vetenskaplig hypotes.2 Att kolesterolkampanjen har kunnat fortsätta beror på att de som författat riktlinjerna för förebyggelse av de kardiovaskulära sjukdomarna har använt vilseledande statistik och ignorerat många misslyckade statinexperiment och talrika motsägelsefulla reporter från oberoende forskare. Vare sig högt totalkolesterol eller högt LDL-kolesterol kan vara orsaken till åderförkalkning eller kardiovaskulär sjukdom. Det finns därför all anledning till att analysera FH individers koagulationssystem i detalj och att sluta med statinbehandlingen. Statinernas nytta är nämligen minimal och förenad med många allvarliga biverkningar.2,3
1. Ravnskov U, de Lorgeril M, Kendrick M, Diamond DM. Inborn coagulation factors are more important cardiovascular risk factors than high LDL-cholesterol in familial hypercholesterolemia. Med Hypotheses 2018;121:60-63. doi: 10.1016/j.mehy.2018.09.019.
2. Ravnskov U, de Lorgeril M, Diamond DM et al. LDL-C does not cause cardiovascular disease: a comprehensive review of current literature. Exp Rev Clin Pharmacol 2018;11: 959-70, DOI: 10.1080/17512433.2018.1519391
3. Diamond DM, Ravnskov U. How statistical deception created the appearance that statins are safe and effective in primary and secondary prevention of cardiovascular disease. Exp Rev Clin Pharmacol 2015;8:201–10.