
Systemisk skleros är en heterogen systemsjukdom med betydande morbiditet och mortalitet, främst på grund av hjärt- och lungengagemang.
Raynauds fenomen, som debuterar i vuxen ålder, är ett vanligt förebud.
Vid diskreta symtom är kontroll av autoantikroppar och kapillärmikroskopi viktiga för diagnos.
Tidig behandling innan irreversibel organskada etablerats är en viktig målsättning.
Behandling ska individanpassas och kan rikta sig mot kärlförändringar i hud och lungcirkulation, hud- och lungfibros, autoimmunitet och gastrointestinalkanalen.
Teamomhändertagande specifikt för systemisk skleros möjliggör bästa vård för många av dessa patienter.
Ett flertal kliniska läkemedelsprövningar pågår för närvarande.
Systemisk skleros, även kallad sklerodermi, är en autoimmun, fibrotiserande systemsjukdom vars låga incidens och prevalens gör att allmänläkare sällan möter eller diagnostiserar sjukdomen. Systemisk skleros angriper såväl huden som inre organ, debuterar ofta smygande och har initialt svårtolkade symtom. Därför kan diagnosen ofta fördröjas. Sjukdomen är heterogen och mortaliteten förhöjd. Betydande framsteg har gjorts avseende patogenes och behandling, och nya läkemedel prövas nu i kontrollerade studier. Två randomiserade studier av autolog stamcellsterapi har visat lovande resultat med halverad mortalitet jämfört med konventionell behandling. Tidig upptäckt av sjukdomsmanifestationer från lungor och andra organ kan ha avgörande betydelse för prognosen, och insatser från fysioterapeut och arbetsterapeut kan förebygga irreversibla handikapp.
Vi vill öka uppmärksamheten på systemisk skleros eftersom tidig diagnos är en förutsättning för ett framgångsrikt omhändertagande av patienterna.
En sällsynt sjukdom med många ansikten
Systemisk skleros karaktäriseras av den unika kombinationen autoimmunitet, kärlskada och fibros i hud och inre organ. Bristande genomblödning underbygger fibros som medför irreversibla skador på lungor, hjärta, njurar, gastrointestinalkanal, hud och leder redan under de första sjukdomsåren. Sjukdomen är ovanlig, med en uppskattad årlig incidens i Sverige på 2/100 000 och prevalens på 30/100 000 [1]. En vårdcentral med ett upptagningsområde på 10 000 individer ser alltså i genomsnitt ett nytt fall vart femte år. Systemisk skleros är 5 gånger vanligare hos kvinnor och debuterar ofta vid 30–50 års ålder.
Systemisk skleros debuterar typiskt med Raynauds fenomen som efterföljs av svullen och sedan förtjockad hud över fingrar och handrygg. Tidig systemisk skleros kan också orsaka ofta svårtolkade symtom såsom klåda, frusenhet, trötthet, avmagring, dysfagi, stelhet och ledvärk. Mindre vanliga debutmanifestationer är mer sjukdomsspecifika och allvarliga; fingergangrän, akut renal kris, lungfibros och pulmonell arteriell hypertension.
Symtomfloran vid etablerad systemisk skleros kan variera stort mellan individer. Andfåddhet är vanligt. I många fall orsakas denna av lungfibros, som förekommer hos en majoritet av patienterna. Vid längre sjukdomsduration är pulmonell arteriell hypertension en vanligare orsak.
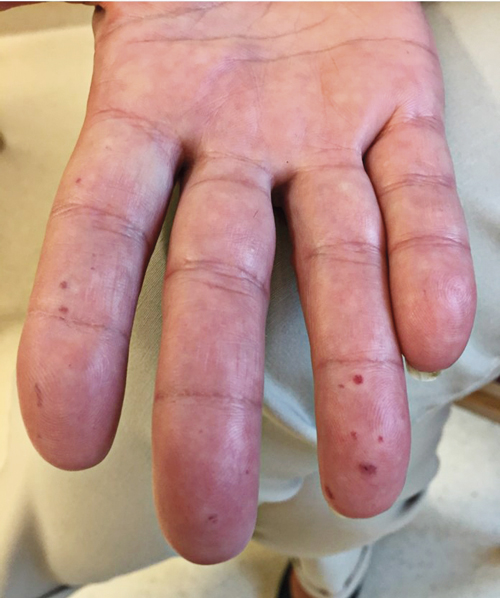
Hudstramhet och klåda orsakas av den initialt inflammerade och sedan fibrotiska huden. Följden blir ofta bestående kontrakturer i små och stora leder. Ansiktsdragen blir strama och gapförmågan minskar. Telangiektasier är vanliga och ibland uttalade, Figur 1.
Sår på fingrar är en frekvent och smärtsam komplikation. Ischemi i fingrar kan också orsaka vävnadsförlust på fingertopparna och utveckling av små gropformade ärr distalt på fingertopparna (pitting scars), Figur 2. Inflammation av leder och krepitationer i senskidor kan också vara framträdande symtom.
Olika gastrointestinala besvär är vanliga vid systemisk skleros. Reflux och/eller patologisk esofagusperistaltik förekommer hos upp till 90 procent. Fekal inkontinens förekommer hos 30 procent och kan vara socialt invalidiserande. Gastrointestinal pseudoobstruktion och blödande ektasier i ventrikelslemhinnan är mindre vanliga, men allvarliga manifestationer. Vanligt hos män är också erektil dysfunktion.
Systemisk skleros delas in i två huvudgrupper: kutant begränsad och kutant diffus. Vid den första begränsas hudförändringarna till ansikte och hudområden distalt om armbåge och knä, medan den senare även angriper proximal hud. Den diffusa sjukdomstypen har ofta ett allvarligare förlopp med tidigt engagemang av inre organ och högre mortalitet [2].
Diagnostik
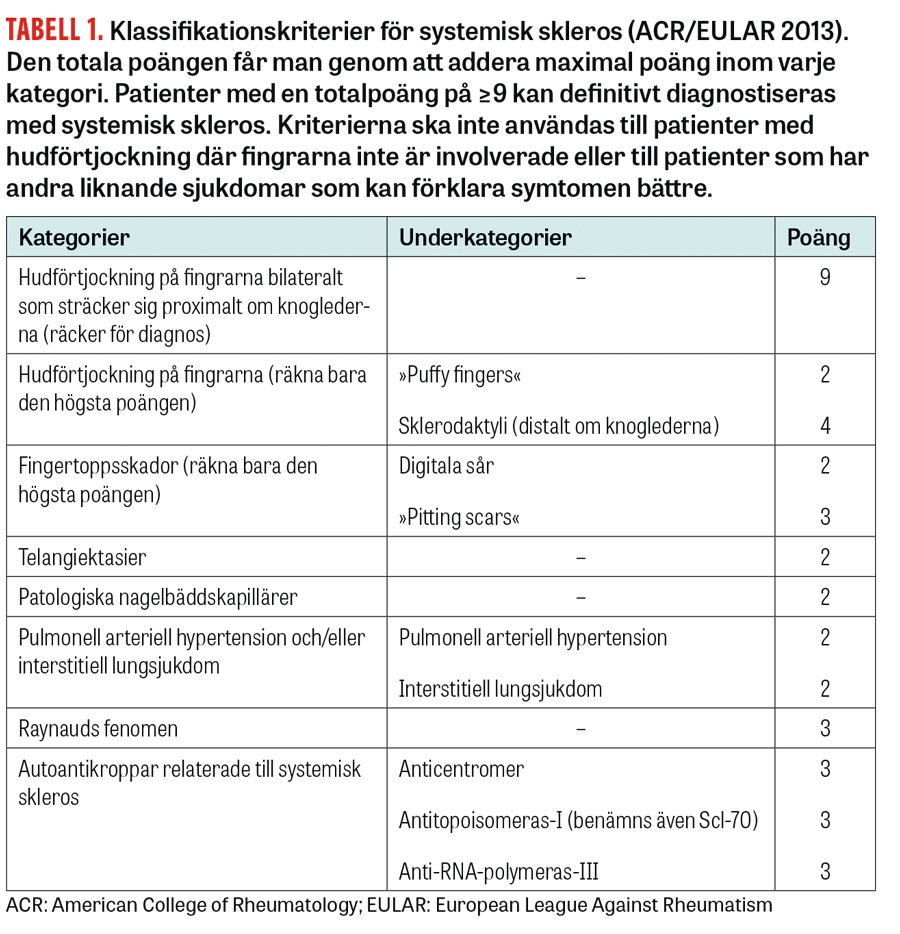
Klassifikationskriterierna uppdaterades 2013 i syfte att underlätta tidig diagnos (Tabell 1) [3]. Kriterierna tar hänsyn till de tre delkomponenterna fibros, kärlpåverkan och autoimmunitet och bygger på ett poängsystem samt baseras på kliniska, radiologiska och immunologiska fynd. Kardinalsymtomen förtjockad hud och Raynauds fenomen liksom tre autoantikroppar specifika för systemisk skleros väger tungt i den algoritm som används. Även patienter helt utan förtjockad hud kan diagnostiseras med systemisk skleros. Många av dessa patienter utvecklar manifest hudförändring först i senare skede.
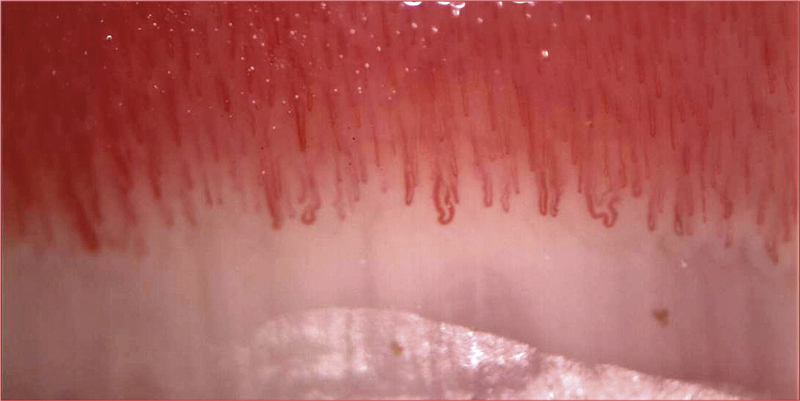
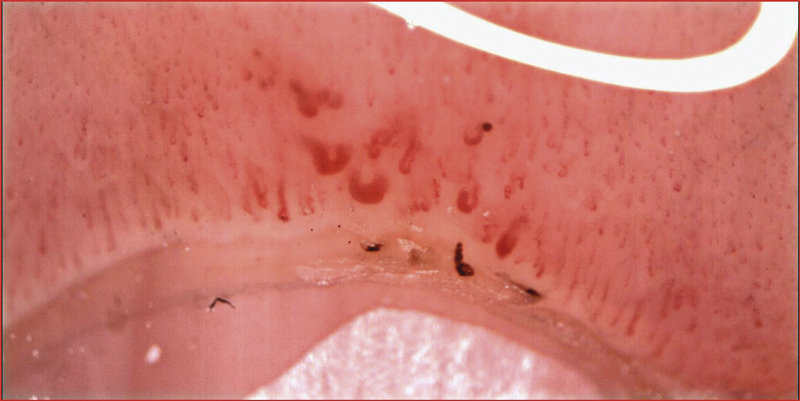
Ovanstående algoritm gäller vid diagnostik av etablerad sjukdom. Men den systemiska sjukdomsdebuten kan föregås av månader eller år med kanske enbart Raynauds fenomen. Patologisk kapillärmikroskopi och cirkulerande autoantikroppar kan då signalera »mycket tidig systemisk skleros«. Kapillärmikroskopi av nagelbädden är etablerad som en enkel bedside-undersökning på dagens reumatologkliniker. Metoden visualiserar de kärl som proximalt om nageln löper parallellt med huden. Typiska fynd är förstorade eller alltför glesa kapillärer, och ibland blödningar (Figur 3 och 4).
Debutsymtomet Raynauds fenomen
Nydebuterat Raynauds fenomen i vuxen ålder är ofta det första förebudet av systemisk skleros. En betydande minoritet (upp till 30 procent) av patienter med sent debuterande Raynauds fenomen kan senare diagnostiseras med reumatisk sjukdom [4]. Differentialdiagnostiska överväganden vid anamnestiska uppgifter om Raynauds fenomen inkluderar [5]
- vibrationsskador
- köldskada
- ändrade rök- och snusvanor
- annan reumatisk sjukdom
- hematologisk malignitet
- läkemedel såsom betablockerare eller sympatomimetika.
Kardinalsymtomet förtjockad hud
Förtjockad hud över fingrar och handrygg är ett ospecifikt fynd som endast i vissa fall orsakas av debuterande systemisk skleros. 2013 års klassifikationskriterier kräver att ett antal ovanliga diagnoser ska exkluderas. Gemensamt för dessa är avsaknad av patologiska nagelbäddskapillärer och av autoantikroppar associerade med systemisk skleros. Gemensamt för ett par av dem är patologisk immunglobulinproduktion [6, 7]. Diagnoserna inkluderar:
- Diabetisk keiroartropati (limited joint mobility, det vill säga begränsad ledrörlighet), en komplikation till både typ 1- och typ 2-diabetes vid vilken i första hand händerna och fingrarna drabbas av stelhet och extensionsdefekter och ibland hudfibros.
- Morfea och linjär sklerodermi, ovanliga fibrotiska hudsjukdomar vars namn och histologi påminner om systemisk skleros men där den kliniska presentationen är begränsad till huden.
- Eosinofil fasciit, där såväl hud som muskelfascia är fibrotiserade. Åtföljs ibland av eosinofili och kan diagnostiseras med biopsi.
- Skleromyxödem, ett ovanligt tillstånd kännetecknat av sklerodaktyli och en hudförtjockning som påminner om den vid systemisk skleros.
- Sklerödem, med proximal hudförtjockning, ibland associerad med mångårig svår diabetes.
- Nefrogen systemisk fibros, ett systemisk fibrotiskt tillstånd inducerat av exponering för gadolinium vid MR-undersökning av patienter med njursvikt.
Sjukdomsspecifika autoantikroppar
Närmare 90 procent av alla patienter med systemisk skleros uppvisar antikroppar mot nukleära antigener (ANA) vid ANA-undersökning. Tre autoantikroppar är av särskild betydelse vid både diagnostik och karakteristik av nydebuterad systemisk skleros. Anticentromerantikroppar påvisas hos patienter med begränsad systemisk skleros och är associerade med sår på fingrar och senare uppkomst av pulmonell arteriell hypertension. Scl-70-antikroppar förekommer hos patienter med diffus systemisk skleros och progressiv lungfibros. RNA-polymeras-III-antikroppar är associerade med förhöjd risk för renal kris och cancer. Den antikroppen undersöks än så länge vid ett fåtal platser i landet.
Etiopatogenes
På både makroskopisk, mikroskopisk och molekylär nivå kännetecknas systemisk skleros av en unik kombination av kärlsjukdom, immunrubbning och fibros [8]. Denna patologi är associerad med komplexa multigenetiska karakteristika och påverkad epigenetik. Sjukdomen uppstår hos genetiskt mottagliga individer till följd av till största delen okända yttre påverkningar. Mycket talar för att autoimmun rubbning i kombination med kärlskada utlöser fibros i hud och andra organ. Därvid omvandlas normala lågaktiva fibroblaster till myofibroblaster, vilka överproducerar extracellulär matrix. Denna fibrotiseringsprocess kan accentuera kärlskada och hypoxi och driva sjukdomen vidare i en negativ spiral. Den har liknats vid en sårläkningsprocess som får pågå ohämmat. Utöver myofibroblaster deltar inte minst endotelceller och immunceller i patogenesen [9]. Cytokiner som TGF-beta och PDGF ökar bildning av myofibroblaster, medan adiponektin från intradermala vita fettceller bromsar omvandlingen.
Djurmodeller av sjukdom liknande systemisk skleros kan induceras med genmanipulation eller tillförsel av toxiska agens, till exempel bleomycin. Dessa förmår dock inte efterlikna alla drag av systemisk skleros. Nyligen har en djurmodell beskrivits där partiell nedreglering av två tillväxtfaktorer alstrar djur med såväl kärlsjukdom, autoimmunitet och fibros. Denna kan få stor betydelse för utveckling av ny terapi [10].
Immunsystemets roll har studerats intensivt ända sedan upptäckten av kärnantikroppar associerade med systemisk skleros och deras specificiteter mot nukleära- och cytoplasmalokaliserade antigener [15]. B-cellsproducerade autoantikroppar, till exempel Scl-70, kan via sin Fc-svans stimulera Fcγ-receptorer, och efter internalisering via TLR7 leda till α-interferonuttryck och inflammation. Aktiverande antikroppar mot angiotensinreceptorer och endotelin-1 kan även bidra till inflammation och kärlskada. B-celler uppvisar minskad produktion av IL-10, vilket resulterar i nedreglering av inflammationsdämpande regulatoriska T-celler. Således deltar B-cellen på ett flertal sätt i patogenesen och borde vara ett relevant mål för riktad terapi.
Cirkulationen i kapillärer och små artärer förändras tidigt. En faktor är endotelin, vars koncentration är förhöjd, vilket leder till vasokonstriktion och även aktiverar fibroblaster. Resultatet blir att endotelceller dör, intiman prolifererar och pericyter vandrar ut i vävnaden och där bidrar till ökad fibros. Även angiogenesen vid systemisk skleros är undertryckt, men likväl har patienterna ofta god läkningsförmåga efter kirurgiska åtgärder.
Samspelet mellan kärlvägg, immunsystem och bindvävsproducerande celler är komplext. Sannolikt har de sjukdomsspecifika patobiologiska processerna vid systemisk skleros redan pågått en längre tid när patienten utvecklat sjukdomsmanifestationer som uppfyller dagens klassifikationskriterier. Det är sannolikt att immunförsvaret är av särskild betydelse vid tidig sjukdom.
Behandlingar
Även om vi i dag identifierat ett flertal prognostiska faktorer för systemisk skleros kan den enskilda patientens prognos inte förutses. De vanligaste relaterade dödsorsakerna är lungfibros, följt av pulmonell arteriell hypertension och njursvikt. Överdödligheten vid systemisk skleros är även i dag betydande, med en genomsnittlig femårsöverlevnad kring 85 procent [11]. Standardiserad mortalitetskvot har uppskattats till 3–4, vilket innebär att dödligheten hos patienter med systemisk skleros är mer än fördubblad jämfört med normalpopulationen. Oftast medför sjukdomen också nedsatt livskvalitet till följd av exempelvis malnutrition eller svårläkta smärtsamma sår [12].
Cyklofosfamid och mykofenolatmofetil har i randomiserade studier visat sig hämma progress av lungfibros associerad till systemisk skleros. Avgörande för dessa läkemedels effekt är att behandling sätts in tidigt i fibrosförloppet eftersom etablerad fibros inte kan läka [13].
Pulmonell arteriell hypertension kan behandlas med viss framgång med fosfodiesteras-5-hämmare (sildenafil, tadalafil) och endotelinreceptorblockerare (bosentan, ambrisentan, macitentan). Prognosen har därmed förbättrats.
Obehandlad akut renal kris leder snabbt till terminal njursvikt, men kan oftast bli reversibel om ACE-hämmare sätts in i tid. Komplikationen är vanligare vid diffus systemisk skleros och oftast förenad med malign hypertoni. Den var tidigare alltid dödlig.
Digitala sår är svårbehandlade. Vi använder i dag vasodilaterande kalciumflödeshämmare, fosfodiesteras-5-hämmare, endotelinreceptorblockerare, nitroglycerinapplikation, intravenös prostacyklinanaloginfusion eller lokala injektioner av botulinumtoxin i handflator. Ortopedisk åtgärd (amputation) kan oftast undvikas [14].
Ektopisk kalcinos subkutant och periartikulärt är svårbehandlad. Vi saknar effektiv medikamentell behandling för denna komplikation. Även ortopediska åtgärder når oftast endast begränsad framgång.
Gastroesofageal reflux kan ofta framgångsrikt behandlas med protonpumpshämmare, men kräver hög dosering livet ut. Malnutrition hanteras bäst tillsammans med dietist och gastroenterolog.
Viktiga allmänna åtgärder förmedlas av funktionsterapeuter. Fysioterapi motverkar kontrakturer och muskelsvaghet. Arbetsterapeuter förmedlar hjälpmedel, till exempel vantar med inbyggda värmeslingor. Kontakt med specialiserade behandlingsteam med erfarenhet av sjukdomen är en ovärderlig tillgång för patienter med systemisk skleros.
Terapi under utveckling
Immunsystemet har en dominerande betydelse i den initiala fasen av systemisk skleros, och en mångfald fas II- och fas III-studier av immunmodulerande terapier har nyligen redovisats eller pågår [14]. Gemensamt för flertalet studier är insikten att behandling bör sättas in tidigt i sjukdomsförloppet för att vara effektiv. En sammanfattning av terapier under utveckling finns i Fakta 2.
Sammanfattning
Trots betydande framsteg rörande patogenes och terapi är systemisk skleros alltjämt en gåtfull och svårbehandlad sjukdom. Tidig diagnos är viktig för att motverka irreversibla skador och av största vikt vid utarbetande av ny terapi. Raynauds fenomen som debuterar i vuxen ålder är ett viktigt förebud och bör följas upp med undersökning av nagelbäddskapillärer och autoantikroppar relaterade till systemisk skleros. Tidig kontakt med specialiserade behandlingsteam är betydelsefull för patienter med systemisk skleros.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Remiss till reumatolog rekommenderas vid misstanke om systemsjukdom vid A + minst en B:
A. Raynauds fenomen med debut i vuxen ålder
B. Övrigt:
- Svåra Raynauds fenomen med eller utan sår
- Svullnad i fingrar
- Telangiektasier
- »Pitting scars« (gropformade ärr)
- Kalcinos
- Autoantikroppar mot centromerer, Scl-70 eller RNA-polymeras-III
- Patologisk kapillärmikroskopi
- Lungfibros
- Pulmonell arteriell hypertension
Observera att detta inte är en »att göra-lista« vid Raynauds fenomen utan enbart varningssignaler för Raynaud sekundärt till systemsjukdom.
Fakta 2. Terapier under utveckling
- Autolog stamcellsterapi har i två omfattande randomiserade studier påvisat en halvering av tvåårsmortaliteten vid diffus systemisk skleros, dock till priset av 5–10 procent akut terapiorsakad mortalitet. Arbete pågår för att optimera denna lovande terapiform.
- Ajulemsyra (lenabasum, tidigare benämnd anabasum) en syntetisk agonist till cannabinoidreceptor 2, har såväl antiinflammatorisk som antifibrotisk verkan i djurmodeller. Detta perorala medel har visat lovande resultat i en mindre fas II-studie och är nu föremål för en större fas III-studie.
- Nintedanib är en tyrosinkinashämmare registrerad för behandling av idiopatisk lungfibros. En nyligen publicerad studie visade positiv effekt också vid lungfibros relaterad till systemisk skleros.
- Pirfenidon är ett antifibrotiskt läkemedel som verkar via TGF-beta och har indikationen idiopatisk lungfibros. Effekten på lungfibros vid systemisk skleros prövas nu i en placebokontrollerad studie.
- Rituximab, en monoklonal antikropp mot B-lymfocyter, har i små randomiserade studier visat potential att bromsa sjukdomen och prövas nu i en fas III-studie.
- Abatacept, CTLA4-Ig, som hämmar effektor-T-celler, har prövats i en fas II-studie som nyligen avslutades.
Referenser
- Andréasson K, Saxne T, Bergknut C, et al. Prevalence and incidence of systemic sclerosis in southern Sweden: population-based data with case ascertainment using the 1980 ARA criteria and the proposed ACR-EULAR classification criteria. Ann Rheum Dis. 2014;73(10):1788-92.
- Proudman SM, Huq M, Stevens W, et al. What have multicentre registries across the world taught us about the disease features of systemic sclerosis? J Scleroderma Relat Disord. 2017;2(3):169-82.
- van den hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum. 2013;65(11):2737-47.
- Pavlov-Dolijanovic SR, Damjanov NS, Vujasinovic Stupar NZ, et al. The value of pattern capillary changes and antibodies to predict the development of systemic sclerosis in patients with primary Raynaud’s phenomenon. Rheumatol Int. 2013;33(12):2967-73.
- Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med. 2016;375(6):556-65.
- Nashel J, Steen V. Scleroderma mimics. Curr Rheumatol Rep. 2012;14(1):39-46.
- Tyndall A, Fistarol S. The differential diagnosis of systemic sclerosis. Curr Opin Rheumatol. 2013;25(6):692-9.
- Fleischmajer R, Perlish JS, Shaw KV, et al. Skin capillary changes in early systemic scleroderma. Electron microscopy and »in vitro« autoradiography with tritiated thymidine. Arch Dermatol. 1976;112(11):1553-7.
- van Laar JM, Farge D, Sont JK, et al; EBMT/EULAR Scleroderma Study Group. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophos-phamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311(24):2490-8.
- Yamashita T, Lakota K, Taniguchi T, et al. An orally-active adiponectin receptor agonist mitigates cutaneous fibrosis, inflammation and microvascular pathology in a murine model of systemic sclerosis. Sci Rep. 2018;8(1):11843.
- Hesselstrand R, Scheja A, Åkesson A. Mortality and causes of death in a Swedish series of systemic sclerosis patients. Ann Rheum Dis. 1998;57(11):682-6.
- Poudel DR, Derk CT. Mortality and survival in systemic sclerosis: a review of recent literature. Curr Opin Rheumatol. 2018;30(6):588-93.
- Volkmann ER, Chung A, Tashkin DP. Manag-ing systemic sclerosis-related interstitial lung disease in the modern treatment era. J Scleroderma Relat Disord. 2017;2(2):72-83.
- Lebedoff N, Frech TM, Shammugam VK, et al. Review of local wound management for scleroderma-associated digital ulcers. J Scleroderma Relat Disord 2018;3(1):66-70.
Summary
Systemic sclerosis is an autoimmune systemic disease with an annual incidence in Sweden of only 20 cases per million and a standardised mortality rate of 3-4. Disease onset is usually preceded by a period with Raynaud’s phenomenon, combined with structurally abnormal nailbed capillaries and accompanied by presence of scleroderma related autoantibodies. The presenting symptoms are skin thickness, puffy fingers, digital ulcers, dysphagia, joint stiffness and pain, and pruritus. Optimal management involves a number of specialists including allied health professionals. Early recognition, diagnosis and treatment are important. The dominating causes of death are cardiopulmonary.