
Typ B-insulinresistenssyndrom (TBIRS) är ett utomordentligt sällsynt autoimmunt tillstånd med polyklonala autoantikroppar riktade mot insulinreceptorn, vilket resulterar i svår och refraktär hyperglykemi och hög mortalitet.
Vi diskuterar här TBIRS utifrån en patient som efter debut av typ 1-diabetes mer än 20-dubblade sitt insulinbehov, men trots detta endast med avsevärd svårighet kunde hålla sitt P-glukos < 40–60 mmol/l.
På misstanke om TBIRS påbörjades en peroral glukokortikoidkur med nedtrappning i avsikt att bryta autoimmun insulinreceptorblockad, vilket gav en omedelbar och dramatisk effekt: på några dagar hade insulinbehovet minskat med 80–90 procent och P-glukos hade stabiliserat sig runt 7–8 mmol/l.
Förekomst av antikroppar mot insulinreceptorn detekterades med immunprecipitation och bindningsanalyser.
Efter en 4 månader lång remission på låg underhållsdos prednisolon recidiverade patienten, varför plasmafereser i omgångar kombinerat med rituximab prövades med tillfällig men övergående god effekt.
Resistens mot insulin blir allt vanligare i takt med att prevalensen av övervikt och fetma ökar i befolkningen. Det anses att bland annat cytokiner som produceras av viscerala adipocyter inducerar insulinresistens genom att interferera med bland annat upptaget av glukos i insulinkänsliga vävnader (till exempel skelettmuskulatur) [1]. Denna »naturliga« eller livsstilsorsakade insulinresistens utgör en mycket viktig och drivande patogenetisk faktor för uppkomsten av prediabetes och manifest typ 2-diabetes hos genetiskt predisponerade individer [1].
Ovanliga former av insulinresistenssyndrom
Det finns emellertid också andra, avsevärt ovanligare former av såväl medfödd som förvärvad insulinresistens associerade med defekter i insulinsignaleringen. En grupp hänför sig till mutationer i insulinreceptorn (till exempel typ A-insulinresistenssyndrom, Donohues syndrom samt Rabson–Mendenhalls syndrom). Utmärkta översiktsartiklar om dessa mycket ovanliga monogena tillstånd finns [2-4].
Typ B-insulinresistenssyndrom (TBIRS) är ett utomordentligt sällsynt autoimmunt tillstånd med förvärvade polyklonala autoantikroppar riktade mot insulinreceptorn, vilket resulterar i svår och refraktär hyperglykemi [5]. Vi beskriver här en ung patient som några månader efter debut av en vanlig och lättskött typ 1-diabetes mer än 20-dubblade sitt insulinbehov men trots detta endast med avsevärd svårighet kunde hålla sitt P-glukos < 40–60 mmol/l.
Fallbeskrivning
Debut av autoimmun diabetes
Patienten, en tidigare väsentligen frisk kvinna av kaukasisk etnicitet, inkom akut 2011 (då 25 år gammal) på grund av nydebuterad diabetes med klassiska prodromalsymtom (polyuri, polydipsi, viktnedgång, trötthet, yrsel, synskärperubbningar) under ett par veckors tid. Patientens farfar hade diabetes av oklar typ, men i övrigt fanns ingen känd hereditet för metabol sjukdom, däremot massiv hereditet för prematur ischemisk hjärtsjukdom. Patienten hade hyperglykemi med icke-fastande P-glukos på cirka 19 mmol/l samt glukosuri 4+, ingen acidos men väl ketonuri 2+. BMI var cirka 36 kg/m2.
Patienten lades in, och man påbörjade sedvanlig fyrdosregim med direktverkande insulinanalog och NPH-insulin (medellångverkande basinsulin). Den glykemiska kontrollen normaliserades snabbt, och patienten hade vid utskrivning en dygnsdos insulin på 28 E (0,3 E/kg). Den glykemiska långtidskontrollen var som förväntat dålig med B-HbA1c på 119 mmol/mol. Serumkoncentrationen av C-peptid låg initialt på hela 1,65 nmol/l. Blodtryck, njurfunktion, U-albumin/kreatininkvot samt retinopatiscreening var utan anmärkning. Patienten hemskrevs med planerad snar uppföljning. Efter utskrivningen anlände svar på analys av autoantikroppar mot β-cellsantigen (GAD-65 > 250 E/ml [ref < 5 E/ml], IA-2 negativt).
Snabb utveckling av svår insulinresistens
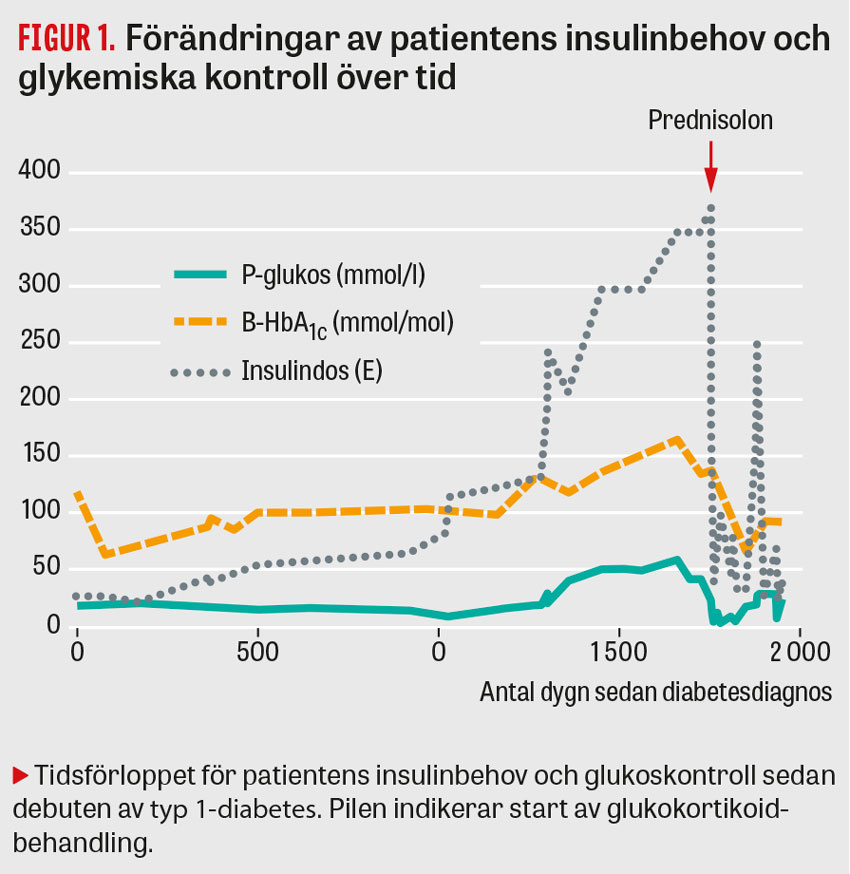
Efter blott några månader med stabil glykemi, väsentligen oförändrade insulindoser och raskt sjunkande HbA1c till 64 mmol/mol märkte patienten att den glykemiska kontrollen sakta men säkert försämrades. Hon ökade sina insulindoser, men det blev allt svårare att hålla en acceptabel glukoskontroll. Någon orsak till denna försämring stod ej att finna. Glykemisk kontroll och insulinbehov över tid framgår av Figur 1.
Situationen blev efter hand alltmer svårhanterbar och patienten hamnade i en utpräglat katabol metabolism med svår hyperglykemi, viktnedgång, uttalad trötthet, ständig huvudvärk, polydipsi och polyuri, vilken även störde hennes nattsömn. Menstruationerna blev oregelbundna och patienten drabbades av återkommande infektioner. Situationen eskalerade ytterligare våren 2014 med kraftigt försämrad glykemisk kontroll (HbA1c > 100 mmol/mol) trots höga insulindoser (> 100 E/dygn). Hon uppvisade inga lipohyper- eller -dystrofier på de varierande injektionsställena. Då patienten var inneliggande noterade diabetessjuksköterskorna god injektionsteknik, och samma dåliga effekt av insulinet blev resultatet då det gavs av vårdpersonal. Periodvis blev situationen så ohållbar att patienten lades in på kliniken några dagar för intravenöst insulindropp i höga doser, vilket initialt gav en bra effekt, som dock snabbt försvann efter utskrivning. Analys av insulinantikroppar utföll negativ vid två tillfällen. Man hade uteslutit monogen diabetes (MODY 1–3). Övrig endokrin utredning (tyreoidea, binjurar, DHEA [dehydroepiandrosteron], GH [tillväxthormon], IGF-1, IGFBP-1 [IGF-bindande protein], proinsulin, glukagon, adiponektin, testosteron, SHBG [sexualhormonbindande globulin], prolaktin, FSH [follikelstimulerande hormon] och LH [luteiniserande hormon]) utföll också helt negativ.
På hemmakliniken, länssjukhuset och region-/universitetssjukhuset hade patienten under cirka fyra års tid i flera omgångar ex juvantibus givits maximala doser av samtliga på marknaden tillgängliga antidiabetiska läkemedelsklasser, såsom metformin, pioglitazon, akarbos, sitagliptin, liraglutid och dapagliflozin i olika kombinationer, med och utan ett flertal olika insulinregimer och -sorter i mycket höga doser, utan någon som helst märkbar effekt. Inte heller sågs någon skillnad då insulinet gavs intramuskulärt eller i kraftigt minskade doser.
Man övervägde även försök med ö-cellstransplantation eller intraperitoneal insulinpump på misstanke om så kallad subkutan insulinresistens, som är en omdiskuterad och kontroversiell entitet [6].
Misstanke om TBIRS
I slutet av 2015 var situationen hopplös och värre än någonsin: Den glykemiska kontrollen var, trots totalt 370 E insulin subkutant per dygn (6,7 E/kg, vid diagnos 28 E/dygn [0,3 E/kg, det vill säga > 20-dubblat insulinbehov]), totalt derangerad med fP-glukos > 35 mmol/l (mätningen visade felkod HHH), P-glukos 50–60 mmol/l och HbA1c 165 mmol/mol. Trots detta var patienten häpnadsväckande opåverkad. Insulinkonsumtionen var nu så stor att patienten hade tvingats köpa ett extra kylskåp enkom för att lagra insulinet. Hennes BMI låg på 22 kg/m2 (före diabetesdebuten 36 kg/m2). Misstanke om TBIRS väcktes nu. Patienten insattes på en peroral steroidkur med nedtrappning i avsikt att bryta den autoimmuna insulinreceptorblockad som karaktäriserar TBIRS. Efter insättandet av 60 mg prednisolon förbättrades situationen snabbt och dramatiskt: inom loppet av 3 dygn kunde man minska insulindosen med 75 procent för att undvika hypoglykemi, en minskning som av samma anledning fortsattes varvid fP-glukos raskt stabiliserade sig kring 6–7 mmol/l på en insulindos om cirka 40 E/dygn. Patientens dagliga dos av prednisolon trappades ned med 10 mg/vecka till underhållsdosen 5 mg × 1. Inga andra stigmata av insulinresistens, såsom hirsutism eller acanthosis nigricans (pigmentering runt hals och armhålor), noterades.
Vid återbesök en månad efter start av kortisonbehandlingen uppgav patienten spontant och med stor glädje en drastiskt förbättrad livskvalitet. Från att tidigare ha varit så extremt trött att hon knappt ens orkat gå och handla eller sköta sin hygien var hon nu mycket piggare. Hon kunde sova ostörd hela nätterna, efter att de senaste 3–4 åren ha plågats av ständigt spring på toaletten på grund av polyuri. Menstruationerna hade också normaliserats, och patienten hade börjat fundera kring barn.
Autoantikroppar mot insulinreceptorn påvisade
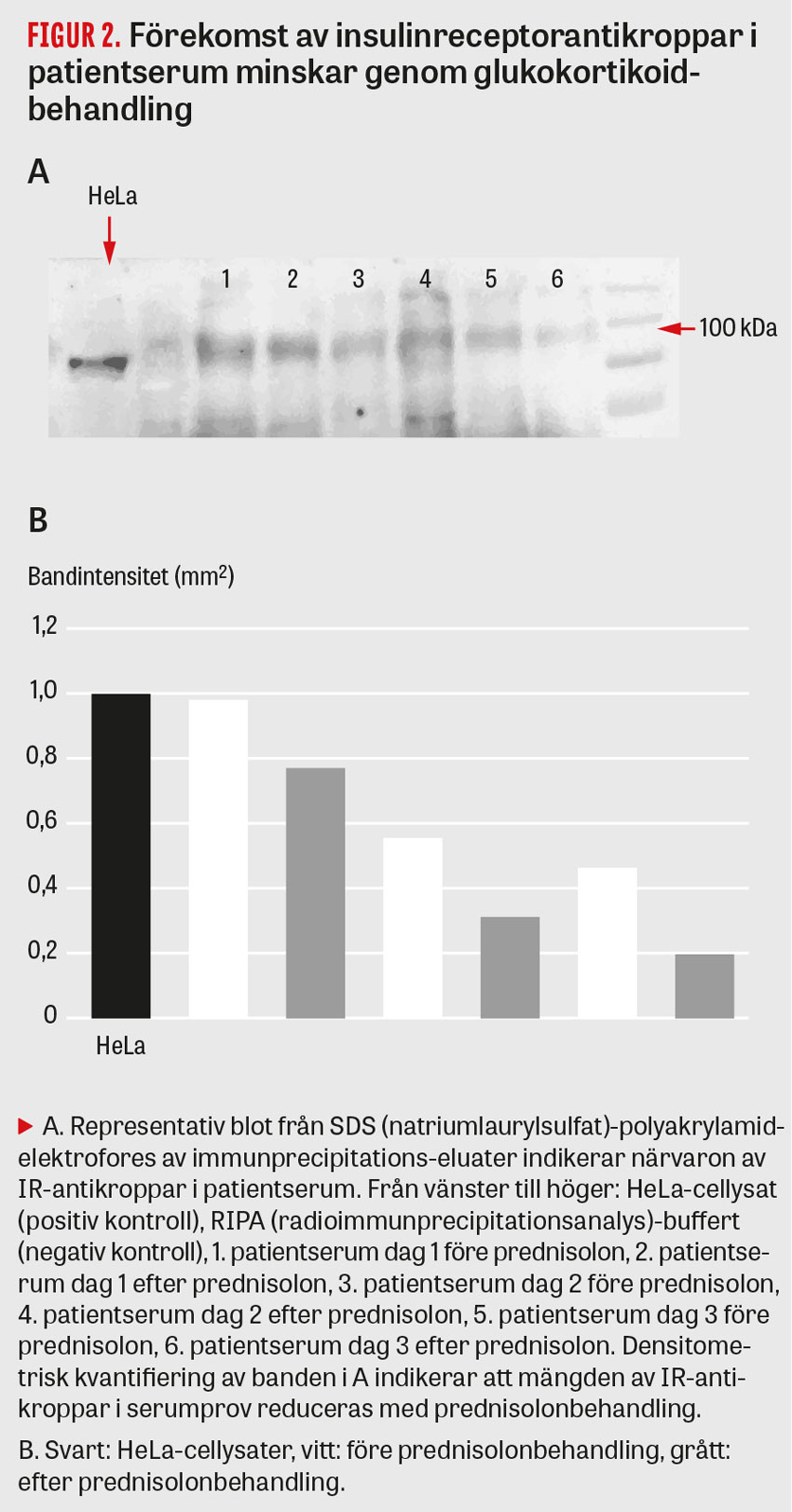
Analyserna gjordes med immunprecipitation [7] respektive bindningsanalys [8]. Resultaten visade närvaro av insulinreceptorantikroppar i patientens serum och en minskning genom glukokortikoidbehandling (Figur 2 A–B). Patientens serum befanns också hämma insulinbindning i adipocyter från en frisk donator, och denna hämning reverserades delvis efter behandling med rituximab och upprepade plasmaferesbehandlingar (Figur 3 A–B).
Recidiv
Ovanstående terapi (med underhållsdosen prednisolon 5 mg/dygn) fungerade alldeles utmärkt i 4 månader. Därefter började, återigen utan känd orsak, glukoskontrollen ånyo att försämras trots ordentliga ökningar i insulindoserna (~ 250 E/dygn) och ökning av dosen prednisolon till 20 mg/dygn. På grund av detta recidiv genomgick patienten en serie om 3 plasmafereser med omedelbar normalisering av glykemi och insulinbehov till »premorbida« nivåer, vilket stärkte misstanken om cirkulerande autoantikroppar mot insulinreceptorn. Hon fortsatte med 20 mg/dygn prednisolon men recidiverade igen blott 4–5 veckor efter plasmafereserna, varför hon gavs en ny serie plasmafereser. Plasmaferesbehandlingen minskade insulinbehovet regelmässigt med cirka 50 procent, en effekt som höll i sig från några dagar upp till ett par veckor. Efter ett tag blev effekten emellertid allt mindre märkbar. Detta gjorde att vi prövade immunadsorption (Globaffinkolonn), som möjliggör behandling av större plasmavolymer, men detta gav ingen förbättring av glukoskontrollen.
Små och kortvariga effekter observerades med rituximab, mykofenolsyra och bortezomib i adekvata doser och behandlingstid. Det har sedan förelegat svårigheter att behålla den något förbättrade glukoskontrollen. Patienten har drabbats av ett flertal ketoacidoser. På grund av svårigheter att hantera det stora insulinbehovet med injektioner har patienten sedermera prövat insulinpump, med något lägre insulindoser, och tidvis blivit avsevärt förbättrad. Behandlingssvårigheterna är i detta fall sannolikt multifaktoriella, och den komplexa diabetessjukdomen med komponenter av såväl typ 1-diabetes som TBIRS är en bidragande orsak.
Diskussion
Patogenes
Vi presenterar här en patient med typ 1-diabetes som utvecklade en extrem insulinresistens till följd av insulinreceptorantikroppar. TBIRS är ett utomordentligt sällsynt tillstånd karaktäriserat av förvärvade antagonistiska autoantikroppar mot insulinreceptorn, vilket kvantitativt blir mest märkbart i skelettmuskulatur, lever och fettväv [5, 11]. Tillståndet är så extremt ovanligt att populationsstudier inte låter sig göras, varför den exakta prevalensen är okänd (en sökning på »type-B insulin resistance« på Pubmed gav endast 75 träffar mellan 1983 och 2019). Som jämförelse kan nämnas att en NIH-klinik i Bethesda, som verkar vara regioninstans för USA:s östkust, mellan 1973 och 2000 endast hanterade 24 patienter [11], det vill säga inte ens ett fall per år. Om man antar en konservativ uppskattning om 50 miljoner invånare i regionen, så skulle det motsvara ett fall vart femte år i Sverige, men stor osäkerhet råder kring denna uppskattning.
TBIRS myntades som en särskild entitet av Jeffrey S Flier när denne för drygt 40 år sedan upptäckte syndromet och dess orsak under sin postdoktorala forskning i C Ronald Kahns laboratorium vid Joslin Diabetes Center i Boston [12, 13].
Graden av insulinresistens som drabbade vår patient bleknar i ljuset av vissa andra publicerade fall, där så mycket intravenöst insulin som 30 000 E/dygn [7] eller till och med 150 000 E/dygn [14] – med andra ord 1,5 liter insulin per dag – inte förmådde påverka hyperglykemin det ringaste. Dessa astronomiska, men helt ineffektiva, doser av insulin låter oss ana potensen och affiniteten hos insulinreceptorantikropparna.
Komorbiditet med andra autoimmunopatier
I litteraturen ses mycket ofta en samsjuklighet med andra autoimmuna tillstånd hos patienter med TBIRS [5, 11]. Systemisk lupus erythematosus, SLE, tycks vara särskilt överrepresenterad hos dessa individer, och det spekuleras om att utbrottet av någon sådan autoimmunopati på något oklart vis även »triggar« uppkomsten av autoantikroppar mot insulinreceptorn. Vår patient hade dock inga antikroppar mot de vanligaste antigenerna (antikroppar mot mitokondrier, nukleära antigener, parietalcellsantigener, citrullin, lever/njurmikrosom typ 1, myeloperoxidas-antineutrofila cytoplasmatiska antigener, proteinas-3-antineutrofila cytoplasmatiska antigener, reumatoid faktor, lösligt leverantigen/leverpankreasantigen, glatt muskelantigen, tyreoperoxidas, vävnadstransglutaminas), eller mot hiv och viral hepatit. Möjligen är det för patienten i vårt fall så att hennes autoimmuna typ 1-diabetes per se utlöst uppkomsten av autoantikroppar också mot insulinreceptorn. Ett fall av TBIRS hos patienter med typ 1-diabetes finns tidigare beskrivet [15]. Patienten har av oklar anledning fått denna receptorimmunopati ovanpå sin autoimmuna typ 1-diabetes och har således i praktiken både typ 1- och typ 2-diabetes.
I en nyligen publicerad fallbeskrivning föreslogs att Helicobacter pylori skulle kunna vara inblandad i etiologin till TBIRS [16]. Vår patient var emellertid seronegativ för denna patogen.
Notabelt är att patienten så sent som i november 2015, nästan 5 år efter diabetesdebuten, hade en C-peptidnivå i serum på 0,68 nmol/l – visserligen under extrem insulinresistens och hyperglykemi, men ändock. År 2014 noterades vid intravenös glukagonbelastning en ökning i serumnivån av C-peptid från 0,37 till 0,73 nmol/l, indikerande stimuleringsbara β-celler. Trots detta indikerar GAD-positiviteten en autoimmun β-cellsdestruktion, varför patienten har en typ 1-liknande diabetes med långsamt förlopp, troligen LADA (latent autoimmun diabetes hos vuxna). Det finns beskrivet att vissa patienter med reguljär typ 1-diabetes kan ha kvarvarande insulinproduktion så länge som 50 år efter debut [17, 18].
Prognos och risk för svår hypoglykemi
Obehandlad är långtidsprognosen generellt för patienter med TBIRS mycket dålig med en tioårsmortalitet på cirka 50 procent [5, 11]. Vanligen beror dödsfallen på komplikationer av intraktabel hypoglykemi. Detta kan tyckas paradoxalt, men har sin förklaring i att autoantikropparna även kan verka agonistiskt på insulinreceptorn [5, 11, 19]. Kronisk svår hyperglykemi och andra manifestationer av grav insulinresistens (såsom dyslipidemi och endoteldysfunktion) innebär å andra sidan välkända risker för angiopati. Vår patient har anamnestiskt haft TBIRS i 7 år, och hon har tung hereditet för prematur ischemisk hjärtsjukdom. Hon är dessutom ung, fertil och barnlös, varför det av flera skäl är utomordentligt viktigt att normalisera hennes insulinkänslighet och glykemi.
Behandling
På grund av att TBIRS är ett så utomordentligt sällsynt tillstånd har behandlingen traditionellt varit empirisk, och ett flertal anekdotiska rapporter med glukokortikoider [7, 14, 20, 21], rituximab [9, 22], cyklofosfamid [8, 10], ciklosporin A [8, 20], azatioprin [10] och plasmaferes [7, 8, 23] har publicerats. Någon evidensbaserad regim har inte funnits, utan många interventioner har skett enligt principen »trial and error«. Nyligen publicerades dock positiva resultat hos en grupp TBIRS-patienter [5, 9, 10]. Hos vår patient hade immunmodulerande behandlingar en initial positiv effekt som dock tyvärr avklingade snabbt.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Honar Dylman och Lars Rönnblom har bidragit med värdefulla råd angående den immunsupprimerande behandlingen.
(uppdaterad 2020-09-24)
Referenser
- Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. 2016;57(12):2099-114.
- Musso C, Cochran E, Moran SA, et al. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore). 2004;83(4):209-22.
- Semple RK, Savage DB, Cochran EK, et al. Genetic syndromes of severe insulin resistance. Endocr Rev. 2011;32(4):498-514.
- Semple RK. EJE Prize 2015: How does insulin resistance arise, and how does it cause disease? Human genetic lessons. Eur J Endocrinol. 2016;174(5):R209-23.
- Willard DL, Stevenson M, Steenkamp D. Type B insulin resistance syndrome. Curr Opin Endocrinol Diabetes Obes. 2016;23(4):318-23.
- Schade DS, Duckworth WC. In search of the subcutaneous-insulin-resistance syndrome. N Engl J Med. 1986;315(3):147-53.
- Page KA, Dejardin S, Kahn CR, et al. A patient with type B insulin resistance syndrome, responsive to immune therapy. Nat Clin Pract Endocrinol Metab. 2007;3(12):835-40.
- Eriksson JW, Bremell T, Eliasson B, et al. Successful treatment with plasmapheresis, cyclophosphamide, and cyclosporin A in type B syndrome of insulin resistance. Case report. Diabetes Care. 1998;21(8):1217-20.
- Manikas ED, Isaac I, Semple RK, et al. Successful treatment of type B insulin resistance with rituximab. J Clin Endocrinol Metab. 2015;100(5):1719-22.
- Malek R, Chong AY, Lupsa BC, et al. Treatment of type B insulin resistance: a novel approach to reduce insulin receptor autoantibodies. J Clin Endocrinol Metab. 2010;95(8):3641-7.
- Arioglu E, Andewelt A, Diabo C, et al. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore). 2002;81(2):87-100.
- Flier JS, Kahn CR, Roth J, et al. Antibodies that impair insulin receptor binding in an unusual diabetic syndrome with severe insulin resistance. Science. 1975;190(4209):63-5.
- Kahn CR, Flier JS, Bar RS, et al. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N Engl J Med. 1976;294(14):739-45.
- Magsino CH Jr, Spencer J. Insulin receptor antibodies and insulin resistance. South Med J. 1999;92(7):717-9.
- Bourron O, Vigouroux C, Halbron M, et al. Association of type B insulin resistance and type 1 diabetes resulting in ketoacidosis. Diabetes Care. 2012;35(2):e4.
- Yang GQ, Li YJ, Dou JT, et al. Type B insulin resistance syndrome with Scleroderma successfully treated with multiple immune suppressants after eradication of Helicobacter pylori infection: a case report. BMC Endocr Disord. 2016;16(1):20.
- Sjöberg S, Ahrén B, Bolinder J. Residual insulin secretion is not coupled to a maintained glucagon response to hypoglycaemia in long-term type 1 diabetes. J Intern Med. 2002;252(4):342-51.
- Yu MG, Keenan HA, Shah HS, et al. Residual β cell function and monogenic variants in long-duration type 1 diabetes patients. J Clin Invest. 2019;129(8):3252-63.
- Bourron O, Caron-Debarle M, Hie M, et al. Type B insulin-resistance syndrome: a cause of reversible autoimmune hypoglycaemia. Lancet. 2014;384(9953):1548.
- Takei M, Ishii H, Kawai Y, et al. Efficacy of oral glucocorticoid and cyclosporine in a case of rituximab-refractory type B insulin resistance syndrome. J Diabetes Investig. 2015;6(6):734-8.
- Mohammedi K, Roussel R, El Dbouni O, et al. Type B insulin resistance syndrome associated with an immune reconstitution inflammatory syndrome in an HIV-infected woman. J Clin Endocrinol Metab. 2011;96(4):E653-7.
- Coll AP, Thomas S, Mufti GJ. Rituximab therapy for the type B syndrome of severe insulin resistance. N Engl J Med. 2004;350(3):310-1.
- Muggeo M, Flier JS, Abrams RA, et al. Treatment by plasma exchange of a patient with autoantibodies to the insulin receptor. N Engl J Med. 1979;300(9):477-80.
- Klubo-Gwiezdzinska J, Lange M, Cochran E, et al. Combined immunosuppressive therapy induces remission in patients with severe type B insulin resistance: a prospective cohort study. Diabetes Care. 2018;41(11):2353-60.
Summary
Type B insulin resistance syndrome (TBIRS) is a very rare autoimmune condition with polyclonal autoantibodies directed against the insulin receptor, which results in severe and refractory hyperglycemia and high mortality. Described here is a patient who, within a few months after the onset of an autoimmune type 1 diabetes, increased her insulin requirements more than 20-fold, and despite this having a considerable difficulty maintaining her P-glucose < 40-60 mmol/L. On suspicion of TBIRS the patient was started on tapering glucocorticoids to overcome the autoimmune insulin receptor blockade, resulting in an immediate and dramatic effect. Within days insulin requirements decreased by 80-90 %, and the P-glucose stabilized around 7-8 mmol/L. The presence of antibodies to the insulin receptor was detected by immunoprecipitation and binding assays. After a 4-month remission on low maintenance dose prednisolone the patient relapsed, which required repeated plasmaphereses with temporarily remarkable effect. Mixed and transient results were seen with rituximab, mycophenolic acid and bortezomib but glycemic control has remained suboptimal. Lack of compliance and recurrent infections may have contributed to this.
En vanligare och ofta missad aspekt på att optimera insulinbehandling hos typ 1 patienter som har (för höga) insulindoser är deras nattsömn, sömnbrist ger enorm påverkan på insulinresistens liksom sömnapneér som bekant.
Därtill har studier visat att den potens som "färskt" insulin som har en intakt köldkedja från produktion till försäljning och förvaring hos patient trots detta finns en variabilitet ofta upp till 50 procent mellan en "over the counter" insulinbehållare jämfört med en annan på apotek i Nordamerika, hur det ser ut i "lilla Sverige" låter jag vara osagt, men dessa faktorer hos t.ex en så svårt sjuk patient får än större genomslag. 1,5 liter insulin om dagen? Den här artikeln kommer jag aldrig att glömma, tack!