![Figur 2. Fylogenetiskt träd baserat på helgenomsekvenser från betavarianten av sars-cov-2 (sekvenstyp B.1.351). Trädet visar släktskap mellan virus från Sverige (rosa) och andra länder (gråskala). De flesta svenska sekvenserna bildar en egen grupp, vilket talar för att de härrör från en enda introduktion som spritts i Sverige snarare än många oberoende importer. Några få oberoende importer ses också längre ner i trädet. Analysen är utförd med den fritt tillgängliga mjukvaran Nextstrain [15] och sekvenser från den internationella databasen GISAID. Figuren utformad av Robert Dyrdak, Karolinska universitetssjukhuset.](https://lakartidningen.se/wp-content/uploads/2021/10/wFig2.jpg)
Storskalig DNA-sekvensering ger nya möjligheter till precisionsmedicin, det vill säga individanpassad handläggning, inom mikrobiologi och infektionsmedicin.
Helgenomsekvensering har tagit över som standardmetod för att bedöma släktskap hos bakterier och virus vid kartläggning av smittspridning.
Helgenomsekvensering möjliggör även utredning av läkemedelsresistens och virulens.
Metagenomisk analys, som ger en bred och förutsättningslös mikrobiologisk diagnostik, förutses komplettera eller ersätta riktade PCR-analyser.
Metagenomisk analys möjliggör även identifiering av nya agens och kartläggning av mikrobiota.
Delning av data och resultat i realtid mellan regioner förbättrar våra förutsättningar att förutse och stoppa utbrott och är ett mål för Genomic Medicine Sweden (GMS).
Genomiska metoder har fått stort genomslag inom klinisk mikrobiologi under de senaste decennierna. Mikrobiella genom är förhållandevis små och av detta skäl enklare att undersöka än humana genom. Inom både bakteriologin och virologin har genomiska metoder till stor del använts för molekylärepidemiologi, men även för molekylär resistenstestning av mikroorganismer. Riktad sekvensering av i förväg definierade eller isolerade mikroorganismer var från början en dominerande metodik, men har successivt kompletterats med metagenomisk diagnostik. Metagenomik syftar till att kartlägga patogena mikroorganismer i ett prov utan att i förväg bestämma vilken/vilka agens analysen riktas mot. Slutligen har intresset även ökat för att kartlägga mikrobiomets, det vill säga normalflorans, betydelse för hälsa och sjukdom.
Precisionsdiagnostik inom bakteriologi och mykologi
Inom klinisk bakteriologi och mykologi har precisionsmedicin använts sedan 1950-talet, i form av fenotypisk art- och resistensbestämning av bakterier och svampar för att kunna rikta antimikrobiell behandling med stor noggrannhet. Molekylära metoder började införas i rutindiagnostik i början av 2000-talet, först i form av olika PCR-metoder för att påvisa resistensgener och sedan även i ökande grad för att påvisa släktskap mellan bakterier i smittspårningssyfte.
Med introduktionen av storskalig massekvensering av bakteriella genom har helgenomsekvensering successivt tagit över från tidigare metoder för molekylär typning i smittspårningssyfte [1] (Figur 1). Dessa metoder har nu använts i minst 5 år för typning av bakterier och mykobakterier på många svenska universitetslaboratorier, vid Folkhälsomyndigheten och på vissa andra laboratorier. Tekniken används i första hand för att kartlägga smittspridning, men ger även möjlighet att identifiera bakteriekloner som är särskilt spridningsbenägna eller som bär gener som gör dem särskilt sjukdomsframkallande, så kallade virulensgener [2]. Denna typ av information kan användas vid handläggningen av patienter, till exempel genom att striktare vårdhygienriktlinjer implementeras i de fall där en mycket spridningsbenägen bakteriestam påträffas. Genomic Medicine Sweden (GMS) är en nationell satsning som syftar till att fler patienter med cancer, sällsynta ärftliga sjukdomar och infektionssjukdomar ska få tillgång till bred genetisk analys för bättre diagnostik och mer individanpassad vård och behandling. Inom ramen för GMS drivs nationellt etablering av storskalig massekvensering, delning av data och resultat i realtid mellan alla regioner och Folkhälsomyndigheten, vilket kan förbättra våra förutsättningar att förutse och stoppa utbrott.
Molekylär resistensbestämning av bakterier, det vill säga påvisande av förvärvade resistensgener eller mutationer i bakteriens arvsmassa, har utvecklats snabbt och förenklas via ett antal publikt tillgängliga dataprogram, såsom Resfinder, AMRfinderplus och CARD (Comprehensive antibiotic resistance database). För bakterier som Staphylococcus aureus och Mycobacterium tuberculosis är korrelationen mellan genomisk och klassisk fenotypisk resistensbestämning mycket god, medan det för andra bakterier som Pseudomonas aeruginosa är en större utmaning att påvisa alla tänkbara förvärvade resistensgener och mutationer [3]. RNA-sekvensering ger möjlighet att komma runt detta problem [4], men är inte väl etablerad än. Denna metod kan kartlägga uttrycket av ett stort antal gener och därmed visa om de är upp- eller nedreglerade. Även för S aureus är fenotypisk resistensbestämning fortfarande den föredragna metoden på grund av lågt pris, snabbhet och enkelhet, men metoden kräver ett framodlat bakterieisolat, medan molekylär resistensbestämning kan göras odlingsoberoende. För en bakterie som M tuberculosis, som tillväxer väldigt långsamt, är molekylär resistensbestämning på väg att få stor betydelse i rutindiagnostiken och den direkta patientvården [5]. Genomisk analys av resistensgener är ännu inte lika utvecklad för svampar, men förväntas introduceras även inom detta område.
Fortfarande är pris och analystid de viktigaste begränsningarna för fortsatt införande av molekylär resistensbestämning och virulensprofilering. Båda metoderna har potential att leda till ökad grad av precision vid behandling av infektioner, men ännu så länge är fenotypiska metoder ofta både snabbare och billigare. Detta kan dock ändras inom kort eftersom det sker snabb utveckling inom DNA-sekvenseringsområdet.
Precisionsdiagnostik inom virologi
För vissa virus och kliniska ställningstaganden är bestämning av virusgenotypen viktig för handläggningen av den enskilda patienten. Ett exempel är behandling av kronisk hepatit C-virusinfektion. Aktuella behandlingsrekommendationer anger att genotypning bör inkluderas i utredningen inför behandlingsstart. Denna genotypning utförs med DNA-sekvensering av en del av virusets arvsmassa. Patientens behandling väljs på basen av bland annat virusets genotyp och leverfibrosstadium. Ett annat aktuellt exempel är sars-cov-2, där helgenomsekvensering blivit något som till och med medier och gemene man diskuterar. Detta beror på att vissa virusvarianter, såsom den sydafrikanska (betavarianten, pangolintyp B.1.351) (Figur 2), har krävt speciella smittskyddsåtgärder. Denna och andra nya varianter av sars-cov-2 kan också komma att kräva anpassade vacciner och vaccinationsscheman.
För många virus som finns med bland agens som omfattas av smittskyddslagen ingår molekylärepidemiologisk undersökning i utredningen av nya fall. Detta gäller bland annat vid mässling (morbillivirus), hepatit A-virus och vissa fall av sars-cov-2. En molekylärepidemiologisk utredning innebär att man inte nöjer sig med att bestämma virusets genotyp, utan även gör en noggrannare utredning av släktskapet mellan patientens virus och andra virus av samma typ. Detta görs ofta med fylogenetiska metoder som gör det möjligt att grafiskt visa virusets släktskap med andra närbesläktade virus i ett släktskapsträd (Figur 2). Dessa molekylärepidemiologiska utredningar gör det möjligt att snabbt kartlägga smittvägar för att göra riktade insatser mot pågående smittspridning i sjukvård och i samhället, det vill säga bryta så kallade smittkluster. RNA-virus, som har en mycket snabb evolution, lämpar sig extra väl för molekylärepidemiologi eftersom särskiljande mutationer uppstår så snabbt att de kan användas för detaljerad kartläggning av smittspridning, ibland ända ner till individnivå.
Resistensbestämning av virus via sekvensering är ett område där precisionsmedicin mycket tidigt infördes i vårdprogram. Hiv är det viktigaste exemplet på detta. Anledningen till att molekylära metoder tidigt kom att användas för resistensbestämning av hiv och andra virus är att det är komplicerat, långsamt och dyrt att använda odlingsbaserade metoder. När det första antivirala läkemedlet för behandling av hivinfektion, zidovudin (AZT, Retrovir), godkändes 1987 stod det snabbt klart att effekten var kortvarig och att detta berodde på att hiv utvecklade resistens mot AZT [6]. Gradvis tillkom fler läkemedel, och kunskap växte fram om att framgångsrik behandling krävde behandling med en cocktail av flera hivläkemedel. Dock har resistens hela tiden kvarstått som ett hot vid läkemedelsbehandling av hivinfektion. Av detta skäl infördes molekylär resistensbestämning via sekvensering i patientvården redan i slutet av 1990-talet, och fanns också med som en viktig komponent i den första svenska behandlingsrekommendationen för hivinfektion, som publicerades 2002. Molekylär resistensbestämning av hiv rekommenderas fortfarande både inför behandlingsstart och vid behandlingssvikt, och tillåter individuellt anpassad antiviral behandling, det vill säga precisionsmedicin [7].
Omfattande sekvenseringsbaserade utredningar i samband med utbrott av exempelvis influensa, sars, mers, ebola och zikavirusinfektion har också bidragit till nya begrepp såsom molekylärepidemiologi och precisionsepidemiologi [8]. Centralt för dessa är ett högupplöst förhållningssätt till att kontrollera sjukdomsspridning, där genomsekvensering kan användas för att fastställa både ursprung, tidpunkt och spridningssätt för viruset.
Metagenomiska applikationer för diagnostik
Med metagenomisk diagnostik avses diagnostik som inte är inriktad på en speciell art, utan där hela det genetiska innehållet i ett prov sekvensbestäms för att sedan med bioinformatisk analys bestämma vilka arter som fanns i provet. 16S rRNA-sekvensering respektive »internal transcribed spacer (ITS) region of nuclear rDNA«-sekvensering av bakterier och svampar är etablerade metoder för odlingsoberoende diagnostik av infektioner [9]. Metoderna är selektiva eftersom de bygger på sekvensering av gener som finns hos samtliga bakterier respektive svampar, men inte hos oss människor. Eftersom 16S rRNA- och ITS-generna är variabla mellan arter så kan sekvenserna användas för att särskilja olika arter. Dessa metoder ger inte information om resistens mot antibiotika och antimykotika, men kan ge artdiagnostik med mycket god upplösning. Tidigare baserades dessa analyser på traditionell så kallad Sangersekvensering, vilket innebar att endast ett fåtal arter per prov kunde påvisas, men nu sker diagnostiken med storskalig massekvensering och bioinformatiska analysflöden som tillåter samtidig identifiering av ett stort antal arter i ett prov. En utmaning är prov med hög förekomst av normalflora, eftersom det kan vara svårt att avgöra orsaken till sjukdom om provet innehåller många olika arter av mikroorganismer. Sådana fall kan kräva multidisciplinär handläggning med nära samarbete mellan klinisk mikrobiologi och behandlande läkare.
Så kallad shotgun-sekvensering är en metod för DNA-sekvensering där ingen PCR-amplifikation av en bestämd målgen sker, utan långa DNA-fragment bryts fysiskt i små fragment som sekvenseras, monteras ihop, analyseras och identifieras med hjälp av olika dataanalysprogram, det vill säga bioinformatisk analys [10]. Metoden kan även påvisa resistensgener och virulensgener, medan det är svårare att associera dessa gener till en särskild mikrobiell art. Shotgun-sekvensering har stor potential att påvisa både kända och potentiellt nya smittämnen och förbättras i takt med att genom från fler organismer kartläggs.
Metagenomiska metoder används också för att karakterisera normalfloran i olika lokaler hos människa och förknippas även med en rad uppmärksammande mikrobiomprojekt som syftat till att utforska icke-odlingsbara mikroorganismer i olika typer av miljöer [11]. Med humant mikrobiom avses framför allt tarmfloran, men även prov från hud, vagina, munhåla och övre luftvägar kan analyseras för påvisande av samtliga mikrobiella gener i provet. Metagenomisk analys är ännu en specialanalys som i dag inte finns tillgänglig på alla kliniska mikrobiologiska laboratorier, men metoden kommer med stor sannolikhet att bli ett viktigt diagnostiskt verktyg för att påvisa sjukdomsframkallande mikroorganismer samt kartlägga den normala mikrobiotan.
Metagenomisk analys införs också gradvis inom den kliniska virologin, där diagnostiken i dag i stor utsträckning baseras på riktade PCR-analyser. PCR-baserad diagnostik har många fördelar: den har hög känslighet, är snabb och går att göra i stor skala. Den stora nackdelen är att målorganismen måste definieras i förväg. Eftersom många infektionssjukdomar har överlappande symtombild gör detta att mer sällsynta eller oväntade mikroorganismer blir svårare att diagnostisera. Virus har inga konserverade genetiska regioner, som bakteriernas 16S- eller svamparnas ITS-regioner, som kan användas för generellt påvisande av virus. Lösningen på detta problem har kommit genom utvecklingen av storskalig sekvensering. I dag kan oselektiv metagenomisk sekvensbestämning av en stor del av arvsmassan i ett prov utföras, till exempel ett luftvägssekret. Bioinformatiska dataprogram används sedan för att jämföra provets gensekvenser med gensekvenser hos kända virus för att se om någon möjlig patogen finns i provet. Denna teknik har använts i ca 15 år för att identifiera nya okända virus, och användes till exempel för att snabbt identifiera sars-cov-2 vid utbrottet i Wuhan. Metoden har också visat sig lämplig för att påvisa oväntade eller förbisedda agens hos svårt sjuka patienter. Ett exempel var ett överraskande fynd av luftvägsviruset coronavirus OC43 hos ett immunsupprimerat barn med fatal encefalit [12]. För att få en känslighet i närheten av PCR-analysens krävs undersökning av ca 10–20 miljoner genfragment i ett enskilt prov. Det kan låta mycket, men den tekniska utvecklingen inom både sekvensering och dataanalys går fort, och därför kan det antas att detta snart kan bli en del av rutindiagnostiken inom mikrobiologin. Tekniken har i dag tagit steget från forskningen till att bli en specialanalys (med en analystid på någon vecka) som erbjuds på ett fåtal laboratorier och används för sällsynta svåra fall. Om analystiden och kostnaderna fortsätter att sjunka i samma takt kommer det snart att vara en teknik med potential att ersätta PCR-diagnostiken på bred front. I så fall kommer mikrobiologisk rutindiagnostik att bli betydligt mer oberoende av att i förväg gissa rätt agens, och det blir även möjligt att identifiera helt nya agens så snart de dyker upp. Utvecklingen av diagnostik genom metagenomisk shot-gunsekvensering har i dag huvudsakligen fokus på virus, eftersom behovet är störst där och tolkningen av resultaten är förhållandevis okomplicerad. Det finns dock inget som principiellt hindrar att diagnostik genom metagenomisk shotgun-sekvensering kan användas för påvisande av patogena bakterier, svampar och parasiter.
Framtida metagenomiska applikationer
Metagenomisk diagnostik av mikrobiomet används ännu knappast alls i rutindiagnostiken. Ett av problemen är att det är svårt att avgöra vad som är ett friskt respektive sjukt mikrobiom. Det är i nuläget oklart vilka metagenomiska applikationer som kan tänkas i framtiden. Vi har valt att ge några exempel på områden där metagenomik kan få en plats framgent.
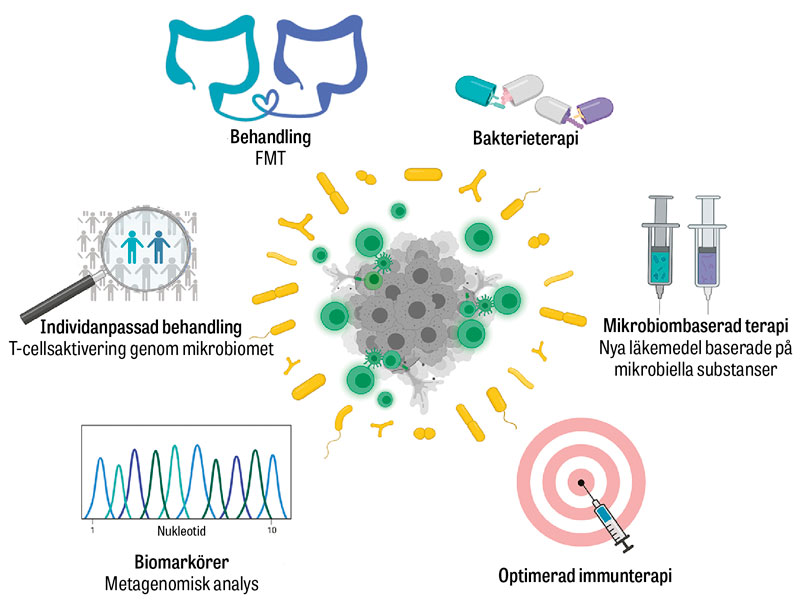
Transplantation av tarmflora från en frisk donator, så kallad fekal mikrobiell transplantation (FMT), har under de senaste åren blivit en viktig del i behandlingen av diarré orsakad av bakterien Clostridioides difficile [13]. Med samma behandlingsstrategi har det visats att bärarskap av resistenta bakterier i tarmen kan minskas [13]. Nya studier har även visat att FMT kan modulera tarmfloran hos patienter med spritt malignt melanom som inte svarat på immunterapi [14]. Donatorn har här varit en patient som blivit botad från sin cancer. En tredjedel av patienterna erhöll regress av tumören när immunterapin sattes in igen efter FMT. Allvarliga biverkningar av immunterapi, såsom kolit, har även kunnat minskas dramatiskt med FMT. Samtliga dessa exempel talar för att metagenomisk analys av tarmfloran kan komma att introduceras inom precisionsmedicinen (Figur 3). Analys av tarmfloran hos vissa cancerpatienter, innan behandling med immunterapi påbörjas, kan bli en viktig del av vårdprogrammet, där prediktion av risken för biverkningar och effekt av behandlingen i så fall är målet. Utöver påvisande av mikroorganismer kan tekniken ge information om det totala antalet resistensgener i tarmen, det så kallade resistomet. Sådan information kan vara av värde för patienter med recidiverande urinvägsinfektioner orsakade av resistenta bakterier.
Sammanfattning
Både helgenomsekvensering av smittämnen och metagenomisk sekvensering har redan fått stort genomslag i infektionsdiagnostiken och kan bidra till ökad precision i handläggningen av patienter. Helgenomsekvensering kan, särskilt om den är kopplad till datadelning enligt utvecklingen inom GMS-projektet, bidra till bättre förutsättningar för att stoppa smittspridning. Helgenomsekvensering av sars-cov-2 har haft stor betydelse vid covid-19-pandemin. Fortfarande finns en stor potential att göra molekylär resistensbestämning av bakterier, virus, och svampar. Metagenomik kan ge möjlighet till odlingsoberoende diagnostik av samtliga smittämnen i ett kliniskt prov. Metoden kan även användas för kartläggning av mikrobiomet och för att förstå dess roll vid både hälsa och sjukdom.
Läs även:
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Molekylära metoder för klinisk mikrobiologisk precisionsdiagnostik
Riktad PCR
Standardmetod för påvisande av virus och svårodlade bakterier och svampar. Specifik för eftersökt art.
Sekvensering av en gen
- 16S-sekvensering och ITS-sekvensering. 16S rRNA- och ITS-generna finns hos alla bakterier respektive svampar, men med olika sekvenser. Oselektiv metod, men fungerar inte för virus. I nuläget begränsad till diagnostik av sterila provmaterial med ett fåtal arter.
- Genotypning. Används för subtypning av vissa smittämnen (till exempel hepatit C-virus)
- Resistensgener. Används för resistensbestämning av till exempel hiv.
Helgenomsekvensering
Sekvensering av hela genomet av en utvald art. Användbart för epidemiologisk typning för smittspårningsutredningar och övervakning samt påvisande av resistens- och virulensgener, till exempel resistenta bakterier och sars-cov-2.
Metagenomik
Sekvensering av all arvsmassa i ett prov. Möjliggör oselektivt påvisande av virus och andra mikroorganismer, inklusive nya okända virus samt kartläggning av normalflora. Kan även appliceras på enstaka gener, till exempel 16S-metagenomik. Har stor potential för fler användningsområden. Det är en bioinformatisk utmaning att tolka de stora mängder data som erhålls.
»Shotgun«-sekvensering
DNA bryts ner i mindre delar, sekvenseras och pusslas därefter ihop med hjälp av bioinformatik. Metod som används för storskalig helgenomsekvensering och metagenomik. Har ersatt traditionell Sangersekvensering för samtliga tillämpningar de senaste 10 åren.
Referenser
- Salipante SJ, SenGupta DJ, Cummings LA, et al. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol. 2015;53(4):1072-9.
- Ny S, Sandegren L, Salemi M, et al. Genome and plasmid diversity of extended-spectrum β-lactamase-producing Escherichia coli ST131 – tracking phylogenetic trajectories with Bayesian inference. Sci Rep. 2019;9(1):10291.
- Ellington MJ, Ekelund O, Aarestrup FM, et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST Subcommittee. Clin Microbiol Infect. 2017;23(1):2-22.
- Li Z, Xu M, Wei H, et al. RNA seq analyses of antibiotic resistance mechanisms in Serratia marcescens. Mol Med Rep. 2019;20(1):745-54.
- Cohen KA, Manson AL, Desjardins CA, et al. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: progress, promise, and challenges. Genome Med. 2019;11:45.
- Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science. 1989;246(4934):1155-8.
- Eriksen J, Carlander C, Albert J, et al. Antiretroviral treatment for HIV infection: Swedish recommendations 2019. Infect Dis (Lond). 2020;52(5):295-329.
- Ladner JT, Grubaugh ND, Pybus OG, et al. Precision epidemiology for infectious disease control. Nat Med. 2019;25:206-11.
- Payne M, Azana R, Hoang LMN. Review of 16S and ITS direct sequencing results for clinical specimens submitted to a reference laboratory. Can J Infect Dis Med Microbiol. 2016;2016:4210129.
- Quince C, Walker AW, Simpson JT, et al. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35(9):833-44.
- Turnbaugh P, Ley R, Hamady M, et al. The human microbiome project. Nature. 2007;449(7164):804-10.
- Morfopoulou S, Brown JR, Davies EG, et al. Human coronavirus OC43 associated with fatal encephalitis. N Engl J Med. 2016;375(5):497-8.
- Walsh D, Gonzalez C, Shannon B, et al. Antimicrobial resistance genes are reduced following administration of investigational microbiota-based therapeutic RBX7455 to individuals with recurrent Clostridioides difficile infection. Open Forum Infect Dis 2020;7(Suppl 1):S16.
- Baruch EN, Youngster I, Ben-Betzzalel G, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371(6529):602-9.
- Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121-3.
Summary
Genomic methods have had a major impact in clinical microbiology in the last decades. Microbial genomes are relatively small and therefore easier to characterise than human genomes. In both bacteriology and in virology, genomic methods have largely been used for molecular epidemiology, but also for molecular resistance testing of microorganisms. Targeted sequencing of predefined or isolated microorganisms was initially a dominant method but has gradually been supplemented with metagenomic diagnostics. Metagenomics aims at mapping all microorganisms – pathogenic as well as apathogenic – in a sample without determining in advance which agent(s) the analysis is targeting. Finally, there is also an increasing interest in mapping the significance of the microbiome, i.e. normal flora, both in health and disease.