Trombocytopeni och samtidig hemolys ska väcka misstanke om trombotisk mikroangiopati (TMA).
Symtomen vid trombotisk trombocytopen purpura (TTP) kan vara diffusa och komma från flera olika organsystem, oftast CNS.
Vid grav trombocytopeni av oklar orsak ska trombocyttransfusioner endast ges efter noggrant övervägande: vid TMA riskerar behandlingen att förvärra tillståndet.
Plasmic-skalan är ett användbart komplement till klinisk bedömning vid misstanke om TTP.
Obehandlad TTP har hög mortalitet. Snabb diagnostik och behandling är avgörande för prognosen.
Plasmaferes med färskfrusen plasma, kortison och rituximab används vid behandling av TTP. Tillägg av kaplacizumab rekommenderas vid allvarlig sjukdomsbild.
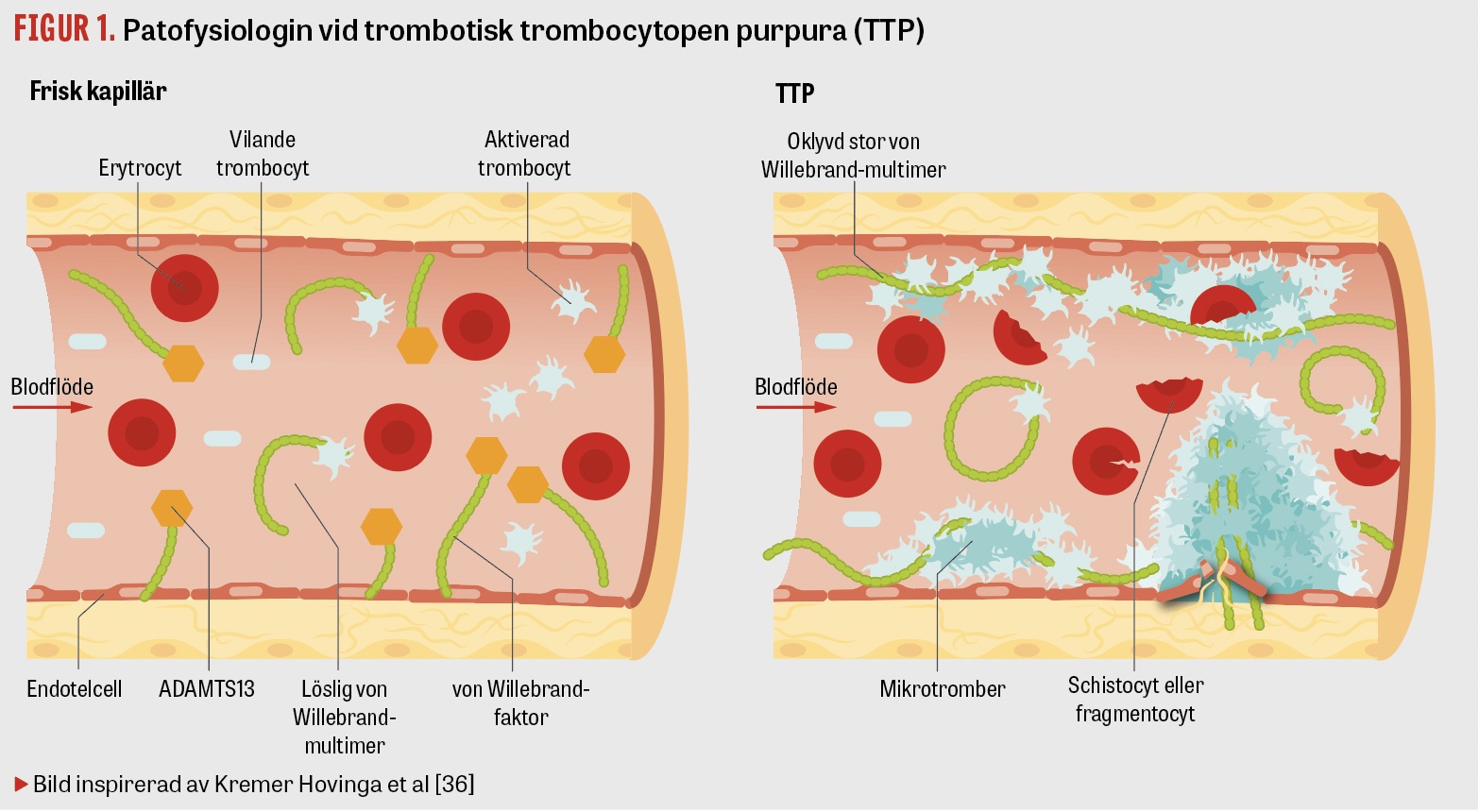
Trombotisk trombocytopen purpura (TTP) är ett slags trombotisk mikroangiopati (TMA), som karakteriseras av mikrotrombotisering i små kapillärer och perifer trombocytkonsumtion. Detta leder till hemolytisk anemi, neurologiska symtom och andra organskador av ischemisk genes [1]. Tillståndet är akut livshotande, men vid tidig behandling med plasmaferes med färskfrusen plasma överlever uppemot 90 procent. Vid obehandlad TTP är däremot mortaliteten drygt 90 procent [2]. Trombocyttranfusioner kan förvärra tillståndet [3].
Tillståndet TTP har tidigare avhandlats i Läkartidningen år 2008 [4, 5] och med fallbeskrivningar år 2018 och 2019 [6, 7]. Det finns dock viktiga nyheter: Plasmic-skalan är ett validerat verktyg och ett bra hjälpmedel i differentialdiagnostiken. Det finns dessutom ny behandling att tillgå: kaplacizumab. En viktig aspekt är också att de laboratorier i Sverige som analyserar enzymet ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif, member 13), det vill säga Skånes universitetssjukhus i Lund och Karolinska universitetssjukhuset i Stockholm, har infört nya metoder som innebär snabbare svarstider vid stark misstanke. 2022 publicerade Svensk förening för hematologi (SFH) riktlinjer för handläggning av TTP, där kaplacizumab lyfts fram i standardbehandlingen. Riktlinjerna finns tillgängliga på www.sfhem.se.
Patientfall
En 74-årig kvinna med Sjögrens syndrom sökte akutmottagningen på grund av huvudvärk, yrsel och allmänt illabefinnande. Hon hade även noterat färskt blod i avföringen. Vitalparametrarna var normala, och patienten var afebril. I provtagning sågs Hb 84 g/l, LPK 5,5 × 10⁹/l och TPK 17 × 10⁹/l. Anemin var normocytär utan tecken på järn-, vitamin B12– eller folatbrist. Elektrolytstatus (Na, K, kreatinin) och CRP var normala. Leverproverna var lätt påverkade med ASAT 1,2 µkat/l, ALAT 1,4 µkat/l, ALP 2,2 µkat/l och bilirubin 48 µmol/l. Man bedömde att det rörde sig om symtomgivande anemi orsakad av misstänkt GI-blödning.
Patienten lades in på kirurgisk vårdavdelning, behandlades med blodtransfusion och genomgick gastroskopi nästa dag, utan patologiska fynd. Patienten försämrades på avdelningen med tilltagande huvudvärk och konfusion. MR hjärna visade bilaterala subaraknoidalblödningar supratentoriellt samt spridda ischemier. Patienten flyttades till strokeenhet, där kompletterande prov bekräftade en hemolysbild: retikulocyter >200 × 10⁹/l, LD 22 µkat/l och haptoglobin omätbart lågt. Trombocyttalet hade sjunkit till 12 × 10⁹/l.
Reumatolog konsulterades, då MR-bilden befanns vara atypisk och man funderade kring eventuell bakomliggande vaskulit. Patienten flyttades till reumatologisk vårdavdelning med misstanke om cerebral systemisk lupus erythematosus. DAT (direkt antiglobulintest) var svagt positivt (1+), vilket ledde till att man satte in högdos steroider. I blodutstryk (erytrocytmorfologi) syntes samtidigt rikligt med schistocyter (fragmenterade erytrocyter).
Hematologkonsult kontaktades och TTP diskuterades som möjlig differentialdiagnos, men ansågs osannolik med tanke på DAT-positiviteten samt frånvaron av njurpåverkan och feber. Provtagningen kompletterades ändå med prov för ADAMTS13. Patienten behandlades för misstänkt cerebral systemisk lupus erythematosus med Sendoxan, systemiska steroider, intravenöst immunoglobulin (IVIG) och upprepade trombocyttransfusioner.
Provsvar dröjde ett antal dagar, men ADAMTS13 visade sig vara uttalat sänkt (0,9 procent av förväntat värde). Kontakt togs med njurmedicin, och plasmaferes inleddes omgående med stark misstanke om TTP. Senare samma kväll insjuknade patienten med medvetandesänkning och vänstersidig hemipares, och akut DT-angiografi bekräftade ocklusion i höger arteria cerebri media. Patienten flyttades till neurointensivvårdsavdelning. Efter daglig plasmaferes i 7 dagar samt behandling med steroider och rituximab normaliserades trombocytvärdet, och patienten förbättrades något kliniskt. Antikroppar mot ADAMTS13 påvisades i hög titer, vilket bekräftade diagnosen förvärvad TTP.
Etiologi
Förvärvad TTP är en autoimmun sjukdom. Antikroppar bildas mot ADAMTS13, vars funktion är att klyva von Willebrand-faktorn, som utsöndras från endotelceller och megakaryocyter. Utan fungerande ADAMTS13 ansamlas stora von Willebrand-faktormultimerer i de små kapillärerna. Dessa binder upp trombocyter och orsakar trombosbildning samt trombocytopeni, då trombocyterna konsumeras (Figur 1).
Den hemolytiska anemin är i typiska fall DAT-negativ och orsakas av att de röda blodkropparna skadas när de passerar mikrotromber i kärlen. Svag DAT-positivitet ses dock vid många tillstånd och förekommer hos upp till 15 procent av sjukhusinlagda patienter [8] och utesluter därför inte TTP.
I blodutstryk ses så kallade schistocyter/fragmentocyter. Ordet schistocyter har sitt ursprung i grekiskans »schistos«, som betyder delad. Schistocyter överstigande 1 procent i perifert blodutstryk är typiska för trombotisk mikroangiopati, men kan förekomma vid ett antal andra tillstånd, t ex membrandefekter, myelofibros eller mekanisk hjärtklaff. Vid TTP är andelen schistocyter i median 6 procent [9], även om stora variationer förekommer, och fall av TTP utan ökning av schistocyter har rapporterats. Man ser även retikulocytos, högt LD och okonjugerat bilirubin samt lågt haptoglobin, i likhet med andra hemolytiska anemier.
Förvärvad TTP är en ovanlig sjukdom. Incidensen beräknas till 1,7 nydiagnostiserade fall per en miljon vuxna invånare och år [10]. Den exakta prevalensen är inte känd, men förvärvad TTP inträffar oftast efter 40 års ålder. Medfödd brist på ADAMTS13 kan ge den primära formen hos barn, men kan även debutera i vuxen ålder, särskilt då behovet av ADAMTS13 ökar, t ex vid graviditet, då koncentrationer av von Willebrand-faktormultimerer successivt ökar [11]. TTP förekommer mer än dubbelt så ofta hos kvinnor som hos män, och i enstaka fall hittas samband med andra autoimmuna sjukdomar, infektioner (hiv eller cytomegalovirus) eller läkemedel [12-14]. Tillstånd eller faktorer som kan misstänkas vara bidragande bör behandlas (eller sättas ut om det gäller läkemedel) parallellt med TTP-specifik behandling. Oftast kan inte någon utlösande orsak till förvärvad TTP identifieras.
Symtom
Klassiskt har man beskrivit en »pentad« bestående av feber, hemolytisk anemi, trombocytopeni, njurpåverkan och neurologiska symtom. Dock uppvisar bara 5 procent av alla patienter samtliga 5 symtom [15]. I en obduktionsstudie av avlidna TTP-patienter sågs mikrotromber i nästan alla organ [16].
Insjuknandet är oftast akut, och symtombilden kan vara högst varierande, från mycket subtila symtom till allvarlig organischemi. Diagnosen måste således misstänkas utifrån konstellationen av laboratorieprover.
Vanligast uppstår symtom från CNS, exempelvis huvudvärk, synbesvär, fokala neurologiska symtom, yrsel, kramper, konfusion och medvetslöshet. Ofta syns en lindrig övergående njurpåverkan.
Mindre än 10 procent av patienterna söker initialt med blödning trots trombocytopeni, som ofta är uttalad [17]. Hjärtpåverkan förekommer också, och troponinläckage betraktas som ett negativt prognostiskt tecken.
Diagnos
Låg ADAMTS13-aktivitet är patognomont för TTP. ADAMTS13-aktivitet <10 procent bekräftar diagnosen hos en patient med trombocytopeni och hemolys. Ett lågt värde kan också ses vid andra trombogena tillstånd, såsom spridd cancer och svår sepsis. De exakta patofysiologiska mekanismerna bakom detta är inte helt klarlagda och sannolikt multifaktoriella, men endotelskada från cytokiner samt en ökad koncentration av von Willebrand-faktorn med sekundär konsumtion av ADAMTS13 har föreslagits [4].
Analysen av ADATMS13 sker vid Karolinska universitetssjukhuset och Skånes universitetssjukhus. Vid stark misstanke om TTP och önskemål om akut analys bör man ta telefonkontakt med koagulationsjour på den specifika analysorten samt laboratoriet. Provsvar som begärs akut erhålls oftast inom 1 dygn. Prov för ADAMTS13 ska tas genom direkt venpunktion (inte via inneliggande kanyl eller port) och innan patienten får plasma.
Provtagningen sker enligt lokala rutiner samt på orten där provet ska analyseras. Information och remisser finns på respektive analysportal (www.analysportalen-labmedicin.skane.se/viewAnalys.asp?Nr=1597 eller www.karolinska.se/pta/klinisk-kemi/adamts13-akut-p–och-adamts13-ak-p–/).
Vid ADAMTS13 <10 procent ska antikroppar mot enzymet analyseras (på det ursprungliga provet). Förekomst av antikroppar talar för förvärvad TTP, medan avsaknad av antikroppar kan tyda på den medfödda formen, och då bör DNA-analys av ADAMTS13-genen genomföras.
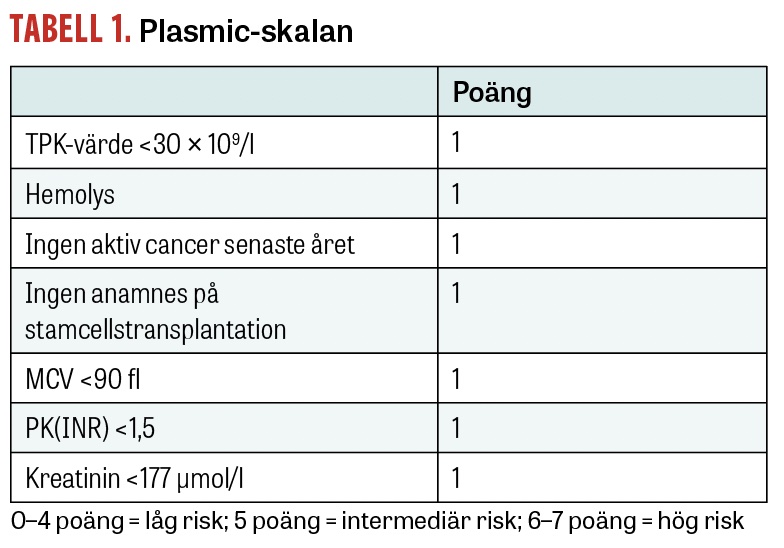
Plasmic-skalan publicerades först 2010 och har sedan validerats i flera ytterligare studier [18, 19]. Se Tabell 1 för vilka parametrar som ingår. Plasmic-skalan är ett användbart komplement till den kliniska bedömningen. Den används för att skilja TTP från andra trombotiska mikroangiopatier och identifiera patienter som kan ha nytta av plasmaferes. Initiering av plasmaferes vid intermediär eller hög risk minskar mortaliteten.
Besläktade tillstånd
Andra trombotiska mikroangiopatier
Medfödd TTP. Medfödd TTP orsakas av mutationer i ADAMTS13-genen, som är autosomalt recessivt nedärvd. Denna form av TTP är klart mer sällsynt än den förvärvade.
Hemolytiskt uremiskt syndrom (HUS). HUS har en liknande klinisk bild som hemolys och trombocytopeni, och det ger ofta en allvarligare njurpåverkan än TTP. HUS förekommer oftare hos barn, men epidemier bland vuxna finns beskrivna. Det utlöses ofta av tarminfektioner med shigatoxinliknande bakterier (enterohemorragiska E coli eller Shigella), där toxinet ger endotelskada. Syndromet kallas också diarréassocierat HUS.
Atypiskt HUS. Atypiskt HUS beror på en överaktivering av komplementsystemet. Det ger liknande klinisk bild som vid HUS, ofta med en uttalad njurpåverkan. Syndromet orsakas oftast av mutationer i gener som kodar för komplementhämmare eller mutationer som leder till att komplementfaktorer blir överaktiva. Uppdaterade riktlinjer från det nationella aHUS-rådet finns tillgängliga på webbplatsen www.njurmed.se.
TMA efter stamcells- eller organtransplantation. Trombotisk mikroangiopati efter stamcells- eller organtransplantation har ofta en mer multifaktoriell patogenes med endoteltoxicitet av läkemedel (ciklosporin), kemoterapi och transplantat kontra värd-sjukdom (GvHD). Som regel uppvisar tillståndet ingen kliniskt signifikant brist på ADAMTS13.
Disseminerad intravasal koagulation (DIC). DIC ses ofta hos svårt sjuka patienter, exempelvis intensivvårdade. Prover visar högt PK, förlängd APTT, förhöjd D-dimer och trombocytopeni. DIC går ibland med hemolytisk anemi, men ofta är andelen schistocyter inom referensintervallet (<0,5 procent) eller strax över [20].
Trombotiska mikroangiopatier hos gravida. Trombotiska mikroangiopatier hos gravida inkluderar preeklampsi och HELLP (hemolysis, elevated liver enzymes, low platelet count syndrome). Vid preeklampsi ses ofta hypertoni och proteinuri. Vid det mer allvarliga HELLP tillkommer även leverpåverkan, trombocytopeni och hemolys. TTP är överrepresenterad vid graviditet och kan då ge leverpåverkan [6]. Vid graviditet kan således differentialdiagnostiken mellan olika trombotiska mikroangiopatier vara svår, och ett nära samarbete med obstetriker rekommenderas.
Andra differentialdiagnoser
Evans syndrom. Vid Evans syndrom ses autoimmun hemolys (DAT-positiv) och immunmedierad trombocytopeni.
Benmärgssjukdom. Benmärgssjukdomar inkluderar exempelvis myelodysplastiskt syndrom (MDS) eller benmärgsinfiltration av lymfom. Dessa kan ge grav trombocytopeni och anemi, men bara i undantagsfall hemolys. Retikulocyttalet är i regel lågt vid dessa tillstånd.
Paroxysmal nokturn hemoglobinuri (PNH). Vid PNH ses DAT-negativ hemolys och tromboser, och hemoglobin utsöndras i urinen och gör den mörkfärgad. PNH brukar i regel inte gå med trombocytopeni.
Behandling
Behandlingen av TTP är komplicerad, tidskrävande, kostsam och ska om möjligt diskuteras med läkare som har erfarenhet av behandling av sjukdomen. Ta gärna kontakt med hematolog på universitetssjukhus eller någon av författarna till SFH:s riktlinjer (2022) för diskussion.
Behandlingen riktas mot tre aspekter av patologin:
- den trombotiska mikroangiopatin (plasmaferes och kaplacizumab)
- autoimmuniteten (kortison och rituximab)
- eventuell bakomliggande sjukdom.
Daglig plasmaferes (plasmabyte) med färskfrusen plasma ska inledas så snart det finns en klinisk misstanke om förvärvad TTP. Detta syftar till att avlägsna autoantikroppar och tillföra nytt ADAMTS13.
Administrering av färskfrusen plasma är indicerad om man inte kan starta plasmaferes omgående. Endast en begränsad mängd färskfrusen plasma kan ges om man inte först tar bort patientens egen plasma med plasmaferes, i annat fall ökar risken för både volym- och proteinbelastning och därmed risken för lungödem.
Man bör undvika trombocyttransfusion.
Kortison ska ges tidigt och i höga doser. Vanlig dos är prednisolon 1 mg/kg. Behandlingen är immunsuppressiv.
Rituximab, en monoklonal antikropp mot CD-20-antigen på B-lymfocyter, ges efter plasmaferes, annars avlägsnas läkemedlet. Standarddos är 375 mg iv/kg/vecka i 4 veckor.
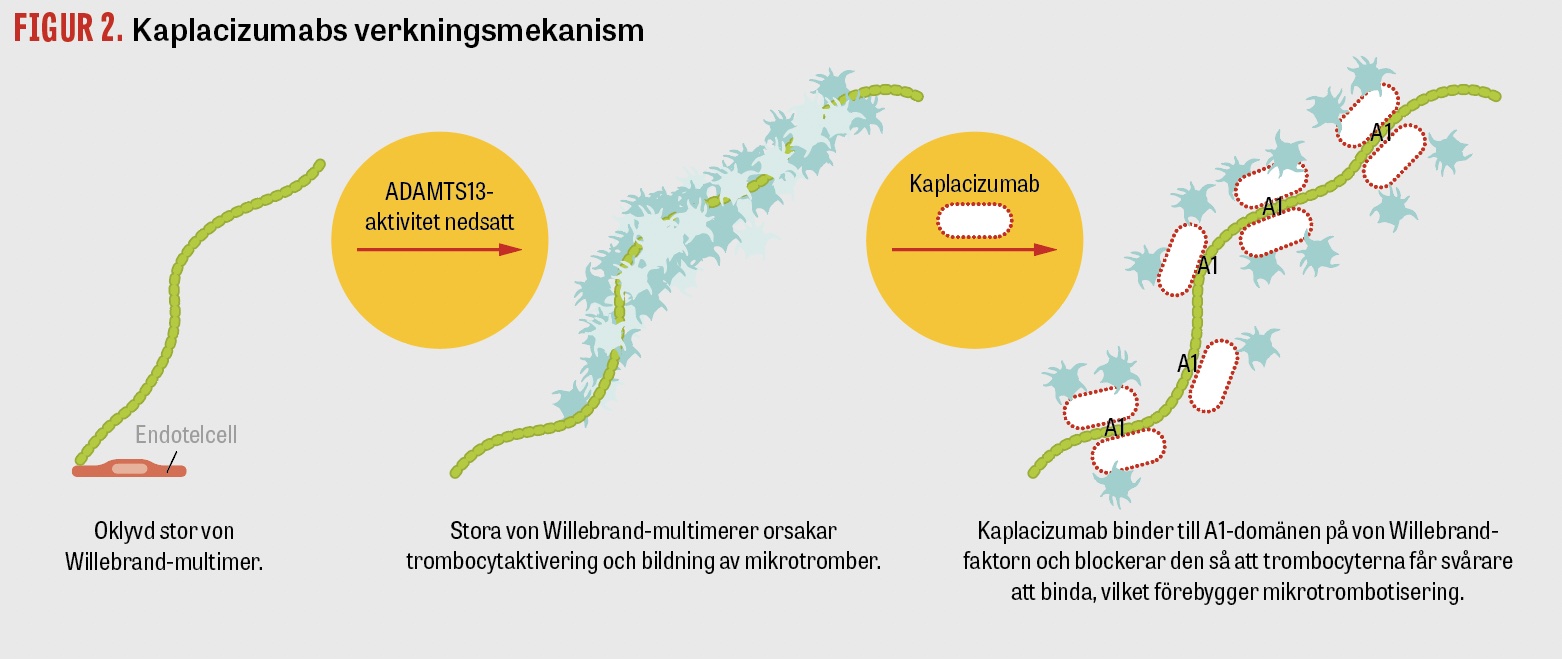
Kaplacizumab (Cablivi) är en monoklonal antikropp (immunglobulinfragment, så kallad nanokropp) mot von Willebrand-faktorn. Kaplacizumab hämmar von Willebrand-faktorns aktivitet genom att hindra dess interaktion med trombocyter och därmed trombocytaggregation samt, i förlängningen, organskada. Läkemedlet har ingen immunsuppressiv effekt (Figur 2). Kaplacizumab ersätter inte plasmaferes, kortison och rituximab, men är ett viktigt komplement [21]. Det finns en ökad blödningsrisk under behandlingstiden, eftersom kaplacizumab även hämmar den interaktion som sker mellan von Willebrand-faktorn och trombocyter inom den normala primära hemostasen. Slemhinneblödningar är vanligt förekommande, men fall av allvarlig blödning har också rapporterats [21], främst hos patienter med blodförtunnande läkemedel. Mätning av von Willebrand-faktor GP1bA-aktivitet kan göras på universitetssjukhus i händelse av en blödningskomplikation. Tillförsel av von Willebrand-faktorkoncentrat kan då övervägas, men bör ske i nära samarbete med koagulationsexpert på grund av det underliggande tillståndet.
Prognos och långtidskomplikationer
Med effektivare behandling har mortaliteten sjunkit från 90 till <15 procent de senaste decennierna [22, 23]. Det finns dock en hög osäkerhet kring mortaliteten i Sverige i dag. Återfall är vanliga och förekommer hos upp till 50 procent av patienterna, med risk för allvarliga komplikationer [24, 25]. Återfallsrisken är högst då ADAMTS13 är <10‒20 procent under remissionsfasen [26, 27]. Behandling med rituximab vid tidiga tecken på sjukdomsaktivitet har visats förebygga återfall [28, 29] och rekommenderas i de svenska nationella riktlinjerna för förvärvad TTP. Därför följs också ADAMTS13 regelbundet efter den akuta behandlingen.
Utöver återfall riskerar TTP-patienter flera långtidskomplikationer, såsom depression, hypertoni, neurokognitiva störningar, stroke, kronisk njursvikt, kardiovaskulära sjukdomar, försämrad livskvalitet och försämrad överlevnad jämfört med referenspopulationer [30-34]. Lågt ADAMTS13 under uppföljningsfasen ökar också risken för stroke [35]. I två kohorter i USA var kardiovaskulära sjukdomar och återfall av TTP de vanligaste dödsorsakerna [34].
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
FAKTA 1. FYND VID UTREDNING AV TTP
- Anemi och hemolys (retikulocytos, högt bilirubin och LD, lågt haptoglobin)
- Hemolysen är oftast DAT-negativ, men DAT-positivitet utesluter inte TTP
- Trombocytopeni
- Fragmenterade erytrocyter (schistocyter)
- Låg ADAMTS13-aktivitet
Referenser
- Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. 2017;130(10):1181-8.
- Page EE, Kremer Hovinga JA, Terrell DR, et al. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. 2017;1(10):590-600.
- Goel R, Ness PM, Takemoto CM, et al. Platelet transfusions in platelet consumptive disorders are associated with arterial thrombosis and in-hospital mortality. Blood. 2015;125(9):1470-6.
- Gøtze JP, Lindblom A, Björk P, et al. ADAMTS13 – aktör och markör vid trombotisk mikroangiopati. Läkartidningen. 2008;105:1092-5.
- Karpman D. Nya rön om EHEC, komplementmutationer och ADAMTS13. Hemolytiskt uremiskt syndrom och trombotisk trombocytopen purpura. Läkartidningen. 2008;105:1096-101.
- Ågren A, Antovic JP, Strandberg K, et al. Tilltagande anemi och trombocytopeni hos gravida kan vara trombotisk trombocytopen purpura. Läkartidningen. 2018;115;EYWT.
- Gustafsson H, Karpman D. Kongenital trombotisk trombocytopen purpura – ett fall med atypisk bild upptäckt i vuxen ålder. Läkartidningen. 2019;116:FF4R.
- Parker V, Tormey CA. The direct antiglobulin test: indications, interpretation, and pitfalls. Arch Pathol Lab Med. 2017;141(2):305-10.
- O’Brien TE, Bowman L, Hong A, et al. Quantification of schistocytes from the peripheral blood smear in thrombotic thrombocytopenic purpura (TTP) compared to non-TTP thrombocytopenic hospitalized patients. Blood. 2018;132(Suppl 1).
- Miesbach W, Menne J, Bommer M, et al. Incidence of acquired thrombotic thrombocytopenic purpura in Germany: a hospital level study. Orphanet J Rare Dis. 2019;14(1):260.
- Coppo P, Veyradier A; French Reference Center for Thrombotic Microangiopathies (CNR-MAT). TTP in the setting of pregnancy: the story still has to be written. J Thromb Haemost. 2020;18(10):2775-7.
- Chiasakul T, Cuker A. Clinical and laboratory diagnosis of TTP: an integrated approach. Hematology Am Soc Hematol Educ Program. 2018;2018(1):530-8.
- Scully M, Cataland S, Coppo P, et al; International Working Group for Thrombotic Thrombocytopenic Purpura. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312-22.
- Sukumar S, Lämmle B, Cataland SR. Thrombotic thrombocytopenic purpura: pathophysiology, diagnosis, and management. J Clin Med. 2021;10(3):536.
- Bugarin-Estrada E, Gómez-De León A, López-García YK, et al. Clinical presentation in thrombotic thrombocytopenic purpura: real-world data from two Mexican institutions. J Clin Apher. 2018;33(6):645-53.
- Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127(7):834-9.
- Griffin D, Al-Nouri ZL, Muthurajah D, et al. First symptoms in patients with thrombotic thrombocytopenic purpura: what are they and when do they occur? Transfusion. 2013;53(1):235-7.
- Bendapudi PK, Hurwitz S, Fry A, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):e157-64.
- Li A, Khalighi PR, Wu Q, et al. External validation of the PLASMIC score: a clinical prediction tool for thrombotic thrombocytopenic purpura diagnosis and treatment. J Thromb Haemost. 2018;16(1):164-9.
- Lesesve JF, Martin M, Banasiak C, et al. Schistocytes in disseminated intravascular coagulation. Int J Lab Hematol. 2014;36(4):439-43.
- Dutt T, Shaw RJ, Stubbs M, et al. Real-world experience with caplacizumab in the management of acute TTP. Blood. 2021;137(13):1731-40.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393-7.
- Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-46.
- Nguyen L, Terrell DR, Duvall D, et al. Complications of plasma exchange in patients treated for thrombotic thrombocytopenic purpura. IV. An additional study of 43 consecutive patients, 2005 to 2008. Transfusion. 2009;49(2):392-4.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol. 2011;86(1):87-9.
- Jin M, Casper TC, Cataland SR, et al. Relationship between ADAMTS13 activity in clinical remission and the risk of TTP relapse. Br J Haematol. 2008;141(5):651-8.
- Peyvandi F, Lavoretano S, Palla R, et al. ADAMTS13 and anti-ADAMTS13 antibodies as markers for recurrence of acquired thrombotic thrombocytopenic purpura during remission. Haematologica. 2008;93(2):232-9.
- Jestin M, Benhamou Y, Schelpe AS, et al; French Thrombotic Microangiopathies Reference Center. Preemptive rituximab prevents long-term relapses in immune-mediated thrombotic thrombocytopenic purpura. Blood. 2018;132(20):2143-53.
- Hie M, Gay J, Galicier L, et al; French Thrombotic Microangiopathies Reference Center. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura. Blood. 2014;124(2):204-10.
- George JN. TTP: long-term outcomes following recovery. Hematology Am Soc Hematol Educ Program. 2018;2018(1):548-52.
- Chaturvedi S, Oluwole O, Cataland S, et al. Post-traumatic stress disorder and depression in survivors of thrombotic thrombocytopenic purpura. Thromb Res. 2017;151:51-6.
- Chaturvedi S, Abbas H, McCrae KR. Increased morbidity during long-term follow-up of survivors of thrombotic thrombocytopenic purpura. Am J Hematol. 2015;90(10):E208.
- Deford CC, Reese JA, Schwartz LH, et al. Multiple major morbidities and increased mortality during long-term follow-up after recovery from thrombotic thrombocytopenic purpura. Blood. 2013;122(12):2023-9; quiz 142.
- Sukumar S, Brodsky M, Hussain S, et al. Cardiovascular disease is a leading cause of mortality among TTP survivors in clinical remission. Blood Adv. 2022;6(4):1264-70.
- Upreti H, Kasmani J, Dane K, et al. Reduced ADAMTS13 activity during TTP remission is associated with stroke in TTP survivors. Blood. 2019;134(13):1037-45.
- Kremer Hovinga JA, Coppo P, Lämmle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers. 2017;3:17020.
Summary
Thrombotic thrombocytopenic purpura (TTP) is a rare life-threatening thrombotic microangiopathy (TMA) characterized by a microangiopathic hemolytic anemia and severe thrombocytopenia, due to platelet consumption. Microthrombi form in small vessels, leading to organ ischemia, most commonly in the central nervous system (CNS). The pathophysiology of TTP is related to a deficiency of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), an enzyme that cleaves the von Willebrand multimer. In the absence of ADAMTS13, the von Willebrand multimer is unfolded into an elongated active form that causes platelet activation and aggregation in arterioles and capillaries. Acquired TTP is caused by autoantibodies against ADAMTS13. The hemolytic anemia is typically DAT-negative and caused by shattering of erythrocytes when passing the microthrombi.
Rapid recognition is crucial for the outcome and to initiate the appropriate treatment. It may take several days to get the test results for ADAMTS13 and when there is a high clinical probability for TTP, plasmapheresis must be initiated pending test results. PLASMIC score can be used in determining the probability of low ADAMTS13 in a hospitalized patient with thrombocytopenia and hemolysis to identify the patients that could benefit from early TTP-specific treatment. First line treatment for acute TTP includes daily plasma exchange, steroids and rituximab. Caplacizumab is an anti-von-Willebrand factor-directed antibody fragment that targets the A1 domain of the von Willebrand factor, thereby inhibiting the interaction between von Willebrand factor multimers and platelets. The treatment has been shown to have beneficial effects when added to standard treatment, without having immunosuppressive effects.