Förvärvad hemofili A bör misstänkas vid oförklarliga blödningar, förlängd APT-tid och normalt PK(INR).
Diagnosen bekräftas av låga nivåer av faktor VIII och förekomst av autoantikroppar mot faktor VIII.
Förvärvad hemofili är en allvarlig sjukdom. Därför är tidig diagnos och behandling avgörande för att minska risken för allvarliga komplikationer och mortalitet.
Förvärvad hemofili A är en sällsynt sjukdom med en årlig incidens på 1–2 fall per miljon, som orsakas av brist på koagulationsfaktor VIII och kan resultera i livshotande blödningar. Sjukdomen orsakas av autoantikroppar mot faktor VIII och är associerad med andra autoimmuna tillstånd och malignitet, men 50 procent av fallen är idiopatiska [1-3]. Inhiberande antikroppar mot faktor VIII resulterar i en dysfunktionell koagulationskaskad och orsakar blödningar, vanligtvis i hud, slemhinnor och muskler. Diagnosen bekräftas genom förlängd aktiverad partiell tromboplastintid (APT-tid), låga faktor VIII-nivåer och förekomst av faktor VIII-antikroppar. Behandlingen omfattar tillförsel av koagulationsfaktorer, immunsuppression och hantering av akuta blödningar [4].
Fallbeskrivning
En 73-årig man med prostatacancer (T2c) som tidigare genomgått radikal prostatektomi inkom med spontana hematom och svullnad i höger arm. 2 månader tidigare hade han genomgått en elektiv ljumskbråcksoperation och postoperativt noterades en svullnad i operationsområdet och ett hematom ned mot skrotum. Hematomevakuering genomfördes utan påvisbar blödningskälla. Patienten hade inte någon pågående antikoagulantiabehandling. Ultraljudsundersökning av armen uteslöt venös trombos och skelettröntgen utföll utan anmärkning. Prov visade Hb 102 g/l, PK(INR) 1,0 och TPK 214 × 109/l, blödningen bedömdes som benign och patienten återgick hem.
Patienten återkom 5 dygn senare då han drabbats av näsblödningar och hade omfattande hematom på hudkostymen. Hb var nu 53 g/l. Det sivade fortfarande vätska från operationsområdet i ljumsken. DT buk visade ett omfattande vänstersidigt retroperitonealt hematom som engagerade m iliacus och m iliopsoas och fortsatte upp mot mjälten utan att en säker blödningskälla kunde påvisas. DT-angiografi av bukkärl visade misstänkt kontrastextravasering från vänster a epigastrica inferior, men någon akut kärlkirurgisk åtgärd bedömdes inte vara aktuell.
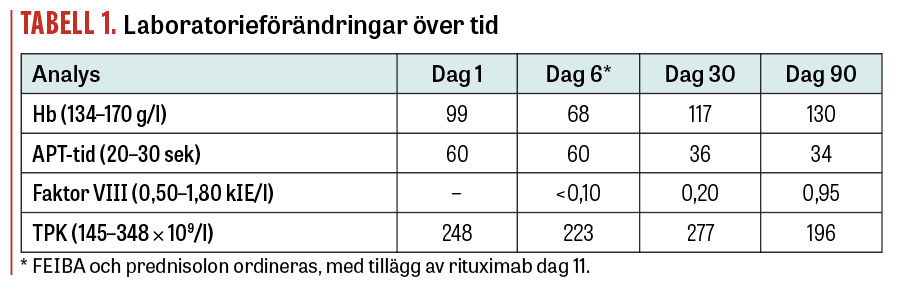
Blödningsutredning inleddes, se Tabell 1. Fibrinogen var 5,6 g/l (referensintervall 2,0–4,2 g/l), faktor VIII-Ak 21 (kBE)/l (<0,5 kBE/l) (BE = Bethesda-enheter), och PK(INR) var vid upprepade tillfällen 1,0 (≤1,2).
Behandling inleddes dag 6 med tranexamsyra, FEIBA (factor VIII inhibitor bypassing activity), erytrocyttransfusioner och prednisolon 50 mg × 1. Dag 11 kompletterades behandlingen med rituximab veckovis under totalt 4 veckor. Näsblödningarna avtog med lokal behandling. Patienten förbättrades och kunde skrivas ut med peroral tranexamsyra och nedtrappning av prednisolon. Faktor VIII, APT-tid och Hb normaliserades, och uppföljningen på hematologmottagningen avslutades efter 3 månader (se Tabell 1).
Diskussion
Vid förvärvad hemofili A bildas IgG-autoantikroppar mot faktor VIII, vilket leder till blödningar och drabbar främst äldre individer [5]. Tidigare blödningar eller antikoagulantiabehandling förekommer vanligtvis inte. Cirka 50 procent av fallen är associerade med autoimmuna sjukdomar (SLE, reumatoid artrit och MS), hematologiska maligniteter eller solida tumörer. Penicillin, interferon och fludarabin är förknippade med tillståndet, och graviditet utgör en riskfaktor, särskilt post partum. Resterande 50 procent av fallen är idiopatiska. Blödningar i hud och muskler är vanligast; melena, hematuri och intrakraniella blödningar förekommer, men det sistnämnda är ovanligt [6, 7].
Förvärvad hemofili bör misstänkas hos patienter med spontana blödningar utan tidigare blödningsanamnes, leversjukdom eller hereditet för blödningssjukdomar med förlängd APT-tid [3]. Laboratorieutredning som bör ingå är blodstatus och differentialräkning för att utesluta malign blodsjukdom. PK(INR) är oftast inom referensintervallet, då det speglar totalaktiviteten av faktor II, VII och X, som är vitamin K-beroende, om det inte finns aktuell antivitamin K-behandling, K-vitaminbrist eller leversjukdom. Låga nivåer av faktor VIII och förekomst av faktor VIII-antikroppar bekräftar diagnosen [8].
I en nordisk multicenterstudie har Lindahl et al sammanfattat det aktuella kunskapsläget om förvärvad hemofili och bekräftar därmed tidigare forskning, särskilt den omfattande studien European acquired haemophilia registry (EACH2) [9], som omfattar en kohort av 181 patienter och betonar sällsyntheten och komplexiteten vid förvärvad hemofili. Sjukdomen kännetecknas av en jämn könsfördelning och en medianålder vid diagnos på 76 år. Den betonar effektiviteten av FEIBA, det vill säga aktiverat protrombinkomplexkoncentrat vid svåra blödningar, vilket står i kontrast till den lägre effektiviteten av rekombinant faktor VIIa. Lindahl et al redovisar 21 procent mortalitet, huvudsakligen sekundär till blödningar och infektioner, vilket understryker behovet av snabbt omhändertagande. Dessa fynd från Norden bidrar till den globala förståelsen av förvärvad hemofili och belyser både gemensamma drag och regionala särdrag i dess manifestation och behandling [1].
Första linjens behandling inkluderar FEIBA, som innehåller faktor II, VII, IX och X. Vid akuta blödningar kan även rekombinant faktor VIIa administreras, och båda dessa läkemedel kan användas antingen som monoterapi eller i kombination. Det finns dock viss risk för trombotiska händelser med dessa behandlingsalternativ. Faktor VIII-koncentrat kan övervägas till patienter med faktor VIII-antikroppar, beroende på antikroppstitern, blödningens svårighetsgrad och lokalisation. Vid höga antikroppstitrar är faktorkoncentrat generellt sett mindre effektivt. Fibrinolyshämmare, såsom tranexamsyra, kan användas vid blödningar och kombineras med faktorkoncentrat men ska undvikas vid hematuri. Plasma, erytrocyt- och trombocyttransfusioner ges vid behov på sedvanlig medicinsk indikation [10].
Immunsuppressiv behandling rekommenderas för att eliminera antikroppar och minska risken för allvarliga blödningar. Prednisolon i kombination med rituximab eller cyklofosfamid rekommenderas i första hand, medan prednisolon som monoterapi kan övervägas. Rituximab bör undvikas som monoterapi på grund av längre tid till remission. Vid utebliven respons på initial behandling kan det behandlingsalternativ som inte använts, det vill säga rituximab eller cyklofosfamid, övervägas som kompletterande behandling [10].
Orsaken till vår patients förvärvade hemofili är inte klarlagd, men tidigare kirurgi kan ha varit en predisponerande faktor. Patofysiologin är dock inte fullständigt känd. Möjligen kan vävnadsskadan ha exponerat autoantigener för immundysreglering, alternativt kan narkosläkemedel ha orsakat en autoimmun reaktion. Symtomen kan uppstå timmar eller dagar efter en operation [11].
Uppföljning efter slutenvård bör initialt ske varannan vecka för att övervaka blödningssymtom, faktor VIII-nivåer och faktor VIII-antikroppar för eventuellt kompletterande behandling. Därefter bör uppföljning fortgå under minst 1 år efter avslutad immunsuppressiv behandling [10].
Läs även författarintervju:
5 frågor till Jolene Johansson
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Referenser
- Lindahl R, Nummi V, Lehtinen AE, et al. Acquired haemophilia A in four north European countries: survey of 181 patients. Br J Haematol. 2023;201(2):326-33.
- Shen M, Wang S, Sessa J, et al. Acquired hemophilia A: a case report. J Pharm Pract. 2020;33(4):562-6.
- Yousphi AS, Bakhtiar A, Cheema MA, et al. Acquired hemophilia A: a rare but potentially fatal bleeding disorder. Cureus. 2019;11(8):e5442.
- Ceresetto JM, Duboscq C, Fondevila C, et al. Acquired haemophilia (acquired factor VIII inhibitor) [artikel på spanska]. Medicina (B Aires). 2015;75(4):231-8.
- Kruse-Jarres R, Kempton CL, Baudo F, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92(7):695-705.
- Rare disease database. Acquired hemophilia. 22 apr 2022. https://rarediseases.org/rare-diseases/acquired-hemophilia/
- Qvist A, Engquist L, Tengborn L. Förvärvad hemofili – förbisedd åkomma med hög morbiditet. Läkartidningen. 2010;107(6):328-32.
- Läkemedelsboken; Astermark J, Berntorp E. Blödningstillstånd. 4 mar 2024. https://lakemedelsboken.se/kapitel/blod/blodningstillstand.html#d3_7
- Knoebl P, Baudo F, Collins P, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;10(4):622-31.
- Nordic Haemophilia Council. Acquired haemophilia. Nordic guidelines. 2020. http://www.nordhemophilia.org/library/Files/PDF-skjol/AcquiredHaemophilia2020.pdf
- Khan UZ, Yang X, Masroor M, et al. Surgery-associated acquired hemophilia A: a report of 2 cases and review of literature. BMC Surg. 2020;20(1):213.
Summary
This case report of a 73-year-old male with bleedings provides insights into the clinical characteristics, diagnosis och treatment of acquired hemophilia A. It’s a dangerous non-hereditary bleeding disorder caused by autoantibodies against coagulation factor VIII, often linked with other autoimmune diseases or malignancies, but it is also often idiopathic. The diagnosis remains a challenge due to its rarity and non-specific symtoms, but should be considered in unexplained bleeding cases with prolonged activated partial thromboplastin time (APTT). Quick diagnosis and treatment can significantly reduce the risk of serious complications and mortality. The long-term prognosis usually depends on the presence of any underlying disease.