De flesta är ense om att
- överväga blodtransfusion/utbytestransfusion vid klinisk försämring hos patienter med vasoocklusiv kris (posttransfusionsmål cirka Hb 90–100 g/l, alternativt reducera HbS till under 50 eller 30 procent)
- höja vårdnivån vid ogynnsam utveckling hos patienter med akut bröstsyndrom
- hematolog bör involveras tidigt i förloppet när patienter med sicklecellsjukdom söker med akuta komplikationer.
Åsikterna går isär om
- huruvida vanliga blodtransfusioner vid akuta komplikationer till sicklecellsjukdom är lika effektiva som utbytestransfusioner, beaktat att man håller sig inom säkerhetsmarginalerna för HbS
- huruvida tidigt andningsstöd kan förbättra utfallet vid akut bröstsyndrom
- hur vården av patienter med sicklecellsjukdom ska organiseras
- vad som ska prioriteras först: trombolys/trombektomi eller blodtransfusioner vid ischemisk stroke.
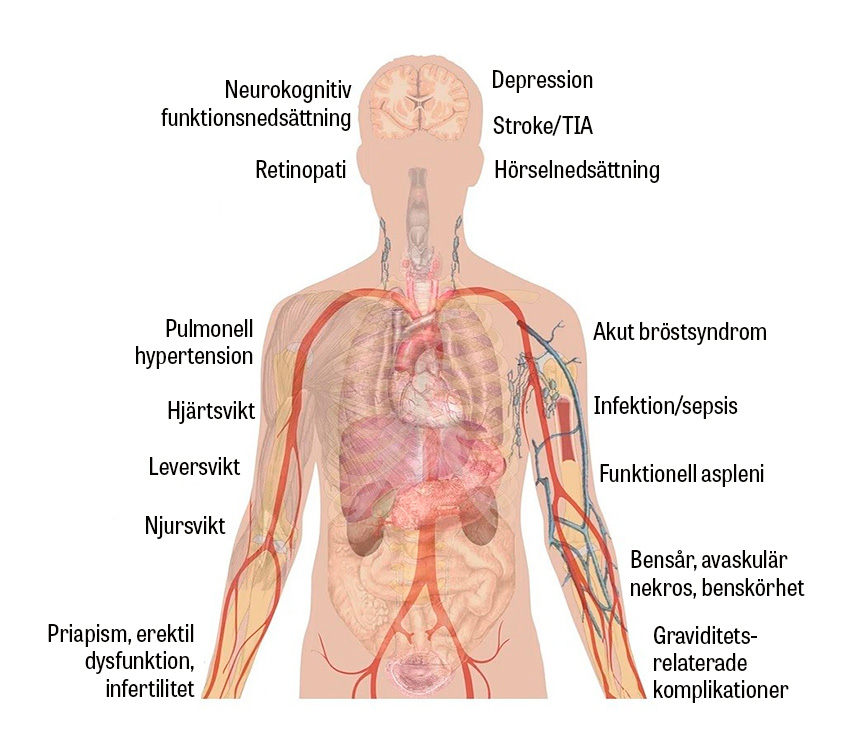
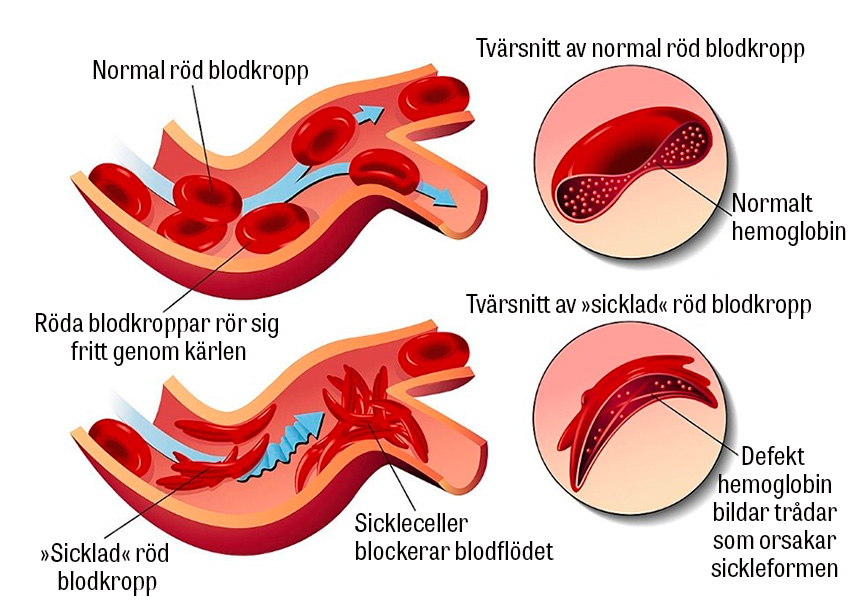
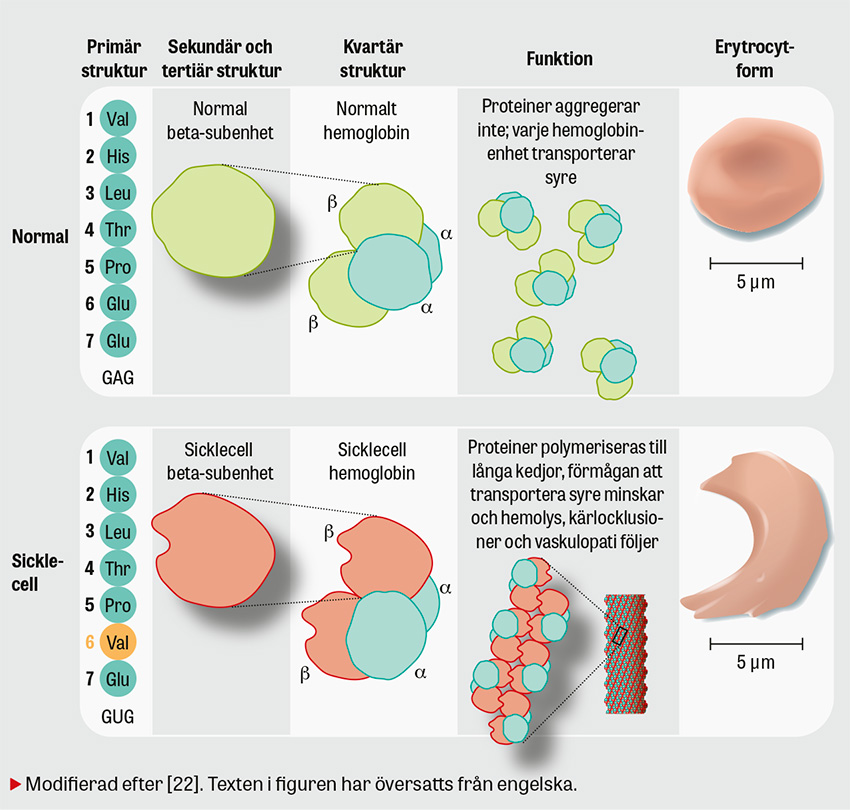
Sicklecellsjukdom är en allvarlig genetisk sjukdom som kännetecknas av att kroppen producerar defekt hemoglobin (HbS). Under syrefattiga förhållanden polymeriseras HbS till långa och stela kedjor i de röda blodkropparna, som blir halvmåne- eller »sickle«-formade. Jämfört med friska röda blodkroppar är de sickleformade cellerna hårdare och böjs inte lika lätt, vilket leder till hemolys, endotelskador och kärlocklusioner med multipla komplikationer som följd.
Sicklecellsjukdom nedärvs autosomalt recessivt. Hos friska individer består hemoglobin av två alfa-globinkedjor och två beta-globinkedjor. Anlagsbärare har en muterad beta-globinkedja medan individer med sjukdomen har ytterligare en mutation. Anlagsbärare är ofta asymtomatiska men kan drabbas av »sickling« under vissa betingelser, som hypoxi, uttorkning, svåra infektioner etc [1].
Över 300 miljoner av världens befolkning är anlagsbärare av sicklecellsjukdom, och varje år föds cirka 515 000 barn med sjukdomen [2]. Sjukdomen är vanligast i Afrika söder om Sahara, Indien, Saudiarabien och Medelhavsländerna. Att vara anlagsbärare, till skillnad från att ha sjukdomen, har evolutionärt varit en överlevnadsfördel i malariaendemiska områden [3]. I Sverige lever cirka 500–600 personer med sjukdomen [4].
Prognosen har förbättrats de senaste åren, mycket tack vare preventivt arbete samt bättre behandling av sjukdomsrelaterade komplikationer. Trots detta är medellivslängden hos patienter med sicklecellsjukdom kraftigt reducerad: med 22 år i USA, och med cirka 15 år om man vårdas på specialistcentrum i London [5]. I Sverige är vården decentraliserad; kvalitetsregister saknas, men ett vårdprogram för vuxna har nyligen publicerats [6].
I denna artikel vill vi gå igenom de vanligaste och farligaste akuta komplikationerna till sicklecellsjukdom hos vuxna och hur dessa handläggs och behandlas.
Behandlingar som kan påverka sjukdomsförloppet
Förstavalsbehandlingen hos symtomatiska patienter med sicklecellsjukdom är hydroxikarbamid, ett cytostatikum som bland annat ökar mängden fetalt hemoglobin, vilket inte påverkas av sicklemutationen. Behandlingen har visat sig minska antalet vasoocklusiva kriser och sjukhusinläggningar, strokerisken hos barn, transfusionsbehov samt mortalitet [7].
Regelbundna blodtransfusioner minskar andelen muterat hemoglobin. Behandlingen har, hos barn, visat sig reducera risken för ischemiska komplikationer såsom stroke [8]. Multipla blodtransfusioner medför dock risk för sekundär hemokromatos och transfusionskomplikationer.
Den enda kurativa behandlingen är allogen stamcellstransplantation. Behandlingen medför risk för komplikationer (avstötningsreaktioner, infertilitet och ökad cancerrisk senare i livet) och en inte oväsentlig mortalitetsrisk [1]. På senare år har genterapi framträtt som alternativ till allogen stamcellstransplantation, och i december 2023 godkände den Europeiska läkemedelsmyndigheten genterapi med Crispr/Cas9-teknik.
Patienter med sicklecellsjukdom drabbas ofta av funktionell aspleni. Utökat vaccinationsskydd mot hepatit A och B, pneumokocker, influensa, meningokocker och sars-cov-2 rekommenderas [9]. Man bör vara uppmärksam på att patienter som inte växt upp i Sverige kan sakna vaccinationer som ingår i det svenska barnvaccinationsprogrammet.
Vasoocklusiv kris
Den vanligaste manifestationen av sicklecellsjukdom är vasoocklusiv kris, som drabbar i princip samtliga patienter med sjukdomen vid ett eller flera tillfällen. Sicklade röda blodkroppar blockerar blodflödet och leder till vävnadshypoxi, vilket i sin tur leder till en inflammatorisk reaktion. Detta resulterar i kraftig smärta, oftast i ryggen, bröstet eller extremiteterna [10].
En majoritet av patienterna som söker akutmottagningen med smärta behöver inneliggande vård. I den akuta situationen ska fokus ligga på smärtlindring samt utredning och behandling av utlösande orsak. Blodstatus, hemolysprov, infektionsprov samt vid behov bastest (stäm av diagnos och eventuell transfusionshistorik med blodcentralen om patienten är ny för regionen) ska tas på akuten. Övrig utredning beror på vilka symtom patienten i övrigt uppvisar. Patienterna ska vara normohydrerade, ha bra syresättning, äta, sova, sköta magen etc, då all form av kroppslig stress riskerar att förvärra sicklingen. Andningsträning med PEP-flöjt förebygger akut bröstsyndrom (se nedan) och ska erbjudas alla inneliggande patienter med sicklecellsjukdom [1].
Mot bakgrund av immundefekten hos patienterna och att infektion ofta är en utlösande orsak till vasoocklusiv kris bör man vara frikostig med antibiotika. Venös tromboembolism är också en vanlig komplikation till sicklecellsjukdom, varför trombosprofylax rekommenderas till alla inneliggande patienter. Blodtransfusioner är sällan nödvändiga och kan potentiellt leda till för hög blodviskositet och försämrad syrgasleverans i vävnaden.
En tredjedel av vuxna patienter med sicklecellsjukdom har daglig smärta, och patienterna känner ofta igen denna smärta vid vasoocklusiv kris. Med det sagt kan patienterna förstås också drabbas av smärt- och sjukdomstillstånd som inte är relaterade till sicklecellsjukdomen, vilket måste beaktas [11].
De flesta patienter med vasoocklusiv kris har redan prövat paracetamol, NSAID och i vissa fall perorala opioider hemma, varför parenteral opioidbehandling som regel är förstahandsval på akuten. Behandling ska påbörjas snarast. Då det är stor variation mellan vilka doser patienterna behöver för att bli adekvat smärtlindrade ska effekt av given behandling följas upp regelbundet. Det hjälper ofta att titta tillbaka på vilka analgetika som givits vid tidigare vårdtillfällen, och ibland finns också behandlingsplaner från patientens hematolog. PCA-pump (patientkontrollerad smärtbehandling) har visat sig vara ett effektivt alternativ [12]. NSAID som tillägg till opioidbehandling rekommenderas också. Hos patienter där ovanstående inte har tillräcklig effekt kan behandling med ketamininfusion eller Catapresan bli aktuellt, ofta efter konsultation med smärtspecialist [13].
Akut bröstsyndrom
Akut bröstsyndrom (acute chest syndrome) är ett allvarligt tillstånd som uppstår på grund av vasoocklusion i lungkärlen hos patienter med sicklecellsjukdom. Detta leder till deoxygenering av hemoglobin och sickling av erytrocyter, vilken orsakar ytterligare vasoocklusion, ischemi och endotelskada. Akut bröstsyndrom kan snabbt progrediera till ett livshotande tillstånd [14]. Cirka 50 procent av patienter med sicklecellsjukdom drabbas någon gång av akut bröstsyndrom, och 10–15 procent av patienterna behöver andningsstöd. Tillståndet har hos vuxna en mortalitet på cirka 3–9 procent, högre ju äldre individen är. Även neurologiska symtom samt låg syresättning förefaller vara ogynsamma prognostiska markörer.
Infiltrat på slätröntgen tillsammans med de kliniska symtomen räcker för att ställa diagnosen, men man ska vara frikostig med datortomografi (DT) vid misstanke om lungemboli. Andra utlösande faktorer är till exempel lunginflammationer, astma och fettembolier. Det är viktigt att alla inneliggande patienter med sicklecellsjukdom kontinuerligt undersöks för andningsbesvär, hosta eller tecken till desaturation för tidig upptäckt och behandling av tillståndet. Precis som vid vasoocklusiv kris ska antimikrobiell behandling ges liberalt och täcka in kapselbärande bakterier och atypiska patogener [15].
Behandlingen, som bör ske i samråd med hematolog, är densamma som vid vasoocklusiv kris men med tätare kontroller, och de flesta patienter kommer att behöva blodtransfusioner. Inte sällan behövs erytraferes (utbytestransfusion), vilken sänker andelen sjuka erytrocyter utan att höja Hb-nivån. Även manuella blodbyten kan övervägas. Ofta behövs blodgruppering och fenotypning av erytrocytantigen (om det inte finns), blodstatus, bastest samt Hb-fraktioner (kvantifiering av HbS) [16]. Svar på Hb-fraktioner ska inte inväntas, utan behandling ska initieras omedelbart.
Bronkdilaterande inhalationer kan bli aktuella hos patienter med känd obstruktiv lungsjukdom eller tecken till akut bronkospasm, och alla patienter ska använda PEP-flöjt. Vid klinisk försämring eller progredierande infiltrat bör patienten skyndsamt flyttas till högre vårdnivå för tätare kontroller samt tillgång till invasivt andningsstöd [17].
Stroke
Sicklecellsjukdom är globalt en av de vanligaste orsakerna till stroke (både ischemisk och hemorragisk) hos barn, och även vuxna patienter har ökad risk att drabbas. Vid 20 års ålder har cirka 10 procent drabbats av klinisk stroke och ännu fler av tysta infarkter [18]. Förekomst av stroke/TIA bör därför alltid övervägas hos patienter med sicklecellsjukdom, oavsett ålder, som söker med neurologiska bortfallssymtom eller svår huvudvärk. Handläggningen, med skyndsam bilddiagnostik (DT skalle med angiografi) är väsentligen densamma som vid stroke hos övriga patienter, men vissa skillnader föreligger.
Patogenesen för stroke hos patienter med sicklecellsjukdom är inte fullt klarlagd, men tros vara en kombination av en ansamling av sicklade erytrocyter, oxidativ endotelskada, förträngningar av blodkärl på grund av onormal vasokonstriktion och ökad aktivitet hos koagulationssystemet. Detta gäller framför allt hos yngre patienter, medan äldre patienter i större utsträckning kan drabbas av stroke av samma orsaker som patienter utan sicklecellsjukdom.
Då man i den akuta situationen sällan har möjlighet att avgöra orsaken till patientens stroke kan både trombolys och trombektomi bli aktuella beroende på NIHSS-poäng (National Institutes of Health stroke scale) och hur lång tid som gått sedan symtomen uppträdde [19]. Patienter med sicklecellsjukdom ska dock också erhålla blodtransfusioner och intravenös vätska. Precis som vid akut bröstsyndrom är erytraferes att föredra, och hyperviskositetsaspekten som omnämns i avsnittet om vasoocklusiv kris måste beaktas [18].
Blödningar är vanligare hos patienter med sicklecellsjukdom än hos patienter utan sicklecellsjukdom, och dessa handläggs på sedvanligt vis. Patienter med sicklecellsjukdom kan drabbas av trombocytopeni, vilket kan förvärra en pågående blödning, och har dessutom ökad risk att drabbas av kärlmalformationer som moyamoya [20].
Mjältsekvestrering
Mjältsekvestrering är en akut, livshotande komplikation som drabbar framför allt barn med sicklecellsjukdom, men även vuxna kan i sällsynta fall drabbas. På grund av sickling minskar cirkulationen i mjälten och en akut ansamling av blod uppstår. Tillståndet progredierar snabbt med fallande Hb och patienten kan inom få timmar drabbas av hypovolem chock. Utan behandling är mortaliteten hög. Tillståndet bör misstänkas vid splenomegali och tecken till cirkulatorisk påverkan i kombination med akut anemi.
Omhändertagandet siktar på att upprätthålla adekvat cirkulation samt att behandla utlösande orsak, ofta en infektion. Både intravenös vätskebehandling och blodtransfusioner krävs som regel, men precis som vid akut bröstsyndrom och stroke är det viktigt att Hb inte stiger för mycket. När tillståndet läker ut återvänder det blod som ansamlats i mjälten till blodbanan, vilket kan leda till hyperviskositet och försämrad perfusion med vasoocklusiv kris eller andra ischemiska komplikationer som följd.
Den stora mjälten kan ge upphov till en sekundär trombocytopeni, varför patienterna ska följas med dagligt blodstatus. Ofta läker tillståndet ut när utlösande orsak behandlats, men i vissa fall kan akut splenektomi bli nödvändig.
En viktig differentialdiagnos till mjältsekvestrering är aplastisk kris, ett tillstånd där benmärgens produktion av erytrocyter minskar kraftigt, vilket också leder till snabbt fallande Hb. Även här är infektion, framför allt parvovirus B19, ofta utlösande orsak, och det snabba Hb-tappet kan leda till sänkt allmäntillstånd och cirkulatorisk påverkan/organsvikt. Tillståndet förbättras ofta spontant efter några dagar [15].
Sammanfattning
Sicklecellsjukdom är en allvarlig kronisk sjukdom med betydande morbiditet och förkortad livslängd. Patienterna kan drabbas av såväl vanliga sjukdomar som komplikationer relaterade till sjukdomen. De sistnämnda kan drabba alla kroppens organsystem. För läkare är det viktigt att snabbt identifiera patienter med sicklecellsjukdom som söker med akuta besvär och skyndsamt påbörja handläggningen.
Läs även författarintervjun med Hedvig Björkman
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Behandling av vasoocklusiv kris
- Smärtstillande behandling ska ges inom 30 minuter från ankomst till akutmottagningen. Målet bör vara VAS 3 eller lägre, och behandlingseffekten ska utvärderas efter senast 30 minuter.
- Blodstatus (Hb, TPK, LPK, retikulocyter, EVF [erytocytvolymfraktion]), hemolysprov (haptoglobin, bilirubin, LD) och infektionsprov (CRP, vid behov mikrobiologisk diagnostik) ska tas vid ankomst till akutmottagningen.
- Patienterna ska vara normohydrerade. Intravenös vätska ges vid behov.
- Syrgastillförsel vid hypoxi (saturationsmål på 95 procent är lämpligt för de flesta patienter) eller vid respiratoriska symtom.
- Blodtransfusion kan övervägas vid Hb <70 g/l eller minskning med 15–20 g/l från patientens habituella nivå.
- Då patienter med sicklecellsjukdom ofta är funktionellt aspleniska ska antimikrobiell behandling som täcker kapselbärande bakterier ges frikostigt vid feber eller andra tecken på infektion.
- Lågmolekylärt heparin i profylaxdos ska ges till alla patienter med inläggningsbehov.
- Vid hypoxi, andningskorrelerade bröstsmärtor och/eller avvikande fynd vid lungauskulation ska radiologi beställas (se avsnitt om akut bröstsyndrom).
- Patienter i behov av inneliggande vård ska använda PEP-flöjt regelbundet.
- Tät uppföljning av patientens tillstånd med till exempel NEWS (National early warning score) behövs under de första dygnen, då tillståndet snabbt kan förvärras.
- Vid utebliven förbättring inom 24–48 timmar eller vid försämring oavsett tidpunkt, överväg behandling med erytraferes.
- Patienterna har behov av lugn på avdelningen och ska helst ligga på enkelsal.
Faktarutan följer rekommendationer från det svenska vårdprogrammet för sicklecellsjukdom [6].
Diagnostiska kriterier för akut bröstsyndrom
Diagnosen akut bröstsyndrom baseras på radiologiska fynd och kliniska symtom. För diagnos ska patienten ha nytillkomna lunginfiltrat på röntgen (slätröntgen, DT) som involverar minst ett lungsegment (infiltratet får ej utgöras av atelektas) samt minst ett av följande symtom:
- Bröstsmärta
- Temperatur över 38,5 °C
- Takypné, väsande andning, rassel, hosta, ökat
andningsarbete - Hypoxemi (>2 procent minskning av SpO2 från normalt läge på rumsluft, PaO2 <8 kPa)
Algoritmen är dock ospecifik och kan också vara diagnostisk för lunginflammation. Patienter med sicklecellsjukdom och nytillkomna luftvägssymtom ska genomgå akut lungröntgen, eftersom det är av stor vikt att påbörja behandling så tidigt som möjligt. Från [14].
Behandling av akut bröstsyndrom
Behandling som vid vasoocklusiv kris med tillägg av nedanstående:
- Blodtransfusion kan bli aktuell redan vid Hb ≤90 g/l eller Hb ≤10 g/l från den habituella nivån, och erytraferes bör övervägas tidigt i förloppet.
- Vid klinisk försämring, ohållbar smärtsituation, progredierande infiltrat eller sjunkande Hb ska överflyttning till högre vårdnivå övervägas.
- Vakna patienter ska använda PEP-flöjt eller motsvarande × 1/h under dygnets vakna timmar.
Faktarutan följer rekommendationer från det svenska vårdprogrammet för sicklecellsjukdom [6].
Behandling av stroke hos patienter med sicklecellsjukdom
- Evidensen kring trombolys/trombektomi är sparsam hos patienter med sicklecellsjukdom, men det är troligt att äldre patienter med andra riskfaktorer för stroke än sicklecellsjukdom gynnas mer än yngre patienter.
- Blodtransfusion, och om möjligt erytraferes, ska genomföras inom 2 timmar från ankomst till sjukhus. I akutskedet bör Hb ligga mellan 85 g/l och 100 g/l.
- På avdelning bör blodprov för kvantifiering av HbS tas, och därefter rekommenderas erytraferes för att sänka andelen HbS <30 procent.
- Hb ska inte överstiga 100 g/l, då ökad blodviskositet kan öka risken för kärlocklusion. Om Hb >100 g/l ska iv vätska ges.
- Blödning är vanligare hos patienter med sicklecellsjukdom än hos bakgrundsbefolkningen och utgör cirka 1/3 av alla strokefall hos denna patientgrupp. Handläggningen är densamma som hos övriga patienter. Dock bör man hos patienter med sicklecellsjukdom vara extra uppmärksam på trombocytopeni, och trombocyttransfusion bör initieras vid <100 × 109/l.
Faktarutan följer rekommendationer från det svenska vårdprogrammet för sicklecellsjukdom [6].
Behandling av mjältsekvestrering
- Intravenös vätska ges för att upprätthålla cirkulation, och akut blodtransfusion är nästan alltid nödvändig. Hb bör dock inte överskrida 80 g/l, då detta ökar risken för komplikationer efter att tillståndet reverserats och de erytrocyter som samlats i mjälten återvänder till cirkulationen.
- Trombocytantal ska kontrolleras, då splenomegali kan leda till trombocytopeni.
- Antimikrobiell behandling (ska också täcka in kapselbärande bakterier) ska ges, då infektion kan vara utlösande orsak.
- I enstaka fall kan akut splenektomi krävas.
Faktarutan följer rekommendationer från det svenska vårdprogrammet för sicklecellsjukdom [6].
Tillvägagångssätt vid manuell utbytestransfusion hos vuxna
Förberedelser
- Venös access (CVK eller två stora PVK).
- Administrera 500 ml NaCl under 15–30 minuter.
- Se till att blodet är uppvärmt före infusion. Använd blodvärmare om tillgänglig.
- Beräkna mängden blod som ska bytas. Hb ≥80 g/l: 5–8 enheter1, Hb 60–79 g/l: 4–6 enheter, Hb <60 g/l: upp till 4 enheter.
Kontroller
- Blodtryck, puls, andningsfrekvens och temperatur kontrolleras regelbundet.
- Patienten ska vara uppkopplad till telemetri och pulsoximeter.
- Ingreppet bör utföras långsammare än vad som beskrivs hos patienter med betydande njur- eller hjärtpåverkan samt vid kardiovaskulär instabilitet.
- Patienten ska hållas euvolem under hela proceduren. Ge ytterligare infusion av NaCl vid behov.
Genomförande
Hb ≥80 g/dl
- Tappa2 1:a enheten samtidigt som den ersätts med 500 ml NaCl.
- Tappa 2:a enheten och transfundera omedelbart 1:a enheten under 30–40 minuter.
- Tappa 3:e enheten och transfundera omedelbart 2:a enheten under 1 timme.
- Tappa 4:e enheten och transfundera omedelbart 3:e enheten under 2 timmar.
Hb 60–79 g/l
- Tappa 1:a enheten och transfundera samtidigt 1:a enheten.
- Tappa 2:a enheten och transfundera därefter 2:a enheten under 1 timme och 3:e och 4:e enheten under 3 timmar.
Hb <60 g/l
- Ge vanlig blodtransfusion till Hb 80–100 g/l (hastighet beroende på kliniskt tillstånd och Hb).
- Utbytestransfusion enligt ovanstående principer kan därefter bli aktuell beroende på patientens kliniska tillstånd.
Utvärdering
- Fullständigt blodstatus, koagulationsprov, HbS3 och elektrolytstatus samt värdera patientens kliniska tillstånd. Om otillräcklig effekt, överväg ytterligare utbyte.
- Vid koagulopati kan administration av plasmaprodukter bli aktuell.
Manuell utbytestransfusion ska endast användas i akuta situationer där erytraferes inte är möjlig, och behandlingen ska alltid ske i samråd med hematologspecialist.
1 En enhet motsvarar cirka 250–350 ml.
2 Tappa 450–500 ml blod under 15–30 minuter.
3 Målet är att sänka HbS-nivån till ≤30 procent samtidigt som Hb hålls nära 100g/l. Klinisk effekt kan dock ses även vid partiellt utbyte.
Källa: oxford-haematology.org.uk
(uppdaterad 2024-07-15)
Referenser
- Inusa BPD, Hsu LL, Kohli N, et al. Sickle cell disease – genetics, pathophysiology, clinical presentation and treatment. Int J Neonatal Screen. 2019;5(2):20.
- GBD 2021 Sickle Cell Disease Collaborators. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000-2021: a systematic analysis from the Global burden of disease study 2021. Lancet Haematol. 2023;10(8):e585-99.
- Bunn HF. The triumph of good over evil: protection by the sickle gene against malaria. Blood. 2013;121(1):20-5.
- Kjellander C, Hernlund E, Ivergård M, et al. Sickle cell disease in Sweden – prevalence and resource use estimated through population-based national registers. Blood. 2021;138(Suppl 1):2040.
- Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: a review. JAMA. 2022;328(1):57-68.
- Svensk förening för hematologi, Arbetsgruppen för sicklecellsjukdom. Vårdprogram för sicklecellsjukdom. 2024. https://www.sfhem.se/riktlinjer
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033-48.
- Howard J. Sickle cell disease: when and how to transfuse. Hematology Am Soc Hematol Educ Program. 2016;2016(1):625-31.
- Socialstyrelsen. Sicklecellsjukdom. 13 jun 2001 [uppdaterat 27 jan 2023]. https://www.socialstyrelsen.se/kunskapsstod-och-regler/omraden/sallsynta-halsotillstand/sicklecellsjukdom/
- Uwaezuoke SN, Ayuk AC, Ndu IK, et al. Vaso-occlusive crisis in sickle cell disease: current paradigm on pain management. J Pain Res. 2018;11:3141-50.
- Darbari DS, Sheehan VA, Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. 2020;105(3):237-246.
- Prestia BM, Ramzan T, Waldron C, et al. Utilization of patient-controlled analgesia reduces length of stay of sickle cell crisis hospitalizations. HCA Healthc J Med. 2021;2(4):303-9.
- Brandow AM, Carroll CP, Creary S, et al. American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv. 2020;4(12):2656-701.
- Friend A, Settelmeyer TP, Girzadas D. Acute chest syndrome. 25 nov 2023. Treasure Island (FL): StatPearls Publishing; 2023.
- Novelli EM, Gladwin MT. Crises in sickle cell disease. Chest. 2016;149(4):1082-93.
- Internet book of critical care (IBCC); Farkas J. Sickle cell acute chest syndrome. 6 jul 2020. https://emcrit.org/ibcc/sickle-chest/
- Howard J, Hart N, Roberts-Harewood M,et al; BCSH Committee. Guideline on the management of acute chest syndrome in sickle cell disease. Br J Haematol. 2015;169(4):492-505.
- DeBaun MR, Jordan LC, King AA, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020;4(8):1554-88.
- Alakbarzade V, Maduakor C, Khan U, et al. Cerebrovascular disease in sickle cell disease. Pract Neurol. 2023;23(2):131-8.
- Baig MU, Bodle J. Thrombolytic therapy. 28 aug 2023. Treasure Island (FL): StatPearls Publishing; 2023.
- Evidence-based management of sickle cell disease. Expert panel report. Bethesda, MD: National Institutes of Health, National Heart, Lung, and Blood Institute (NHLBI); 2014.
- Tan MI. Cell and molecular biology for diagnostic and therapeutic technology. J Phys Conf Ser. 2016;694:012001.
Summary
Sickle cell disease is a genetic disorder affecting hundreds of thousands of people worldwide. This article will focus on the most common and dangerous acute complications to sickle cell disease. Vaso-occlusive crisis is the most common manifestation of sickle cell disease. Rapid pain relief and treating the underlying cause are cornerstones of the treatment. Acute chest syndrome is the most common cause for intensive care unit admission and death among adult patients with sickle cell disease. The condition is treated as vaso-occlusive crises, but patients often need blood transfusions and respiratory support. Patients with sickle cell disease have an increased risk of stroke. Treatment follows the usual guidelines for stroke with the addition of blood- or exchange transfusions. Splenic sequestration is a rare but acute and life-threatening complication, most commonly seen in children. The condition can quickly lead to hypovolemic shock and the treatment aims to maintain adequate circulation and to treat the underlying cause.