De vanligaste orsakerna till högt ferritin är överkonsumtion av alkohol, metabolt syndrom, akut eller kronisk inflammation och järnöverskott (primärt eller sekundärt).
Hos patienter med högt ferritin och normal transferrinmättnad utvärderas alkoholkonsumtion, förekomst av metabolt syndrom och tecken till inflammation eller infektion.
Vid förhöjd transferrinmättnad kontrolleras HFE-genotyp för att fastställa eventuell primär (hereditär) hemokromatos, eller så undersöker man om det finns tecken till sekundär hemokromatos.
Vid normal HFE-genotyp måste ett eventuellt järnöverskott i levern bekräftas med magnetresonanstomografi innan behandling med venesectio påbörjas.
Ferritin i serum är en av de mest efterfrågade analyserna i både primär- och slutenvård, och ofta ses förhöjda värden [1]. Det finns ingen konsensus kring den exakta övre normalvärdesgränsen, men vanligen anges den till 200 µg/l för kvinnor och 300 µg/l för män. Ett lågt ferritinvärde har en hög sensitivitet och specificitet för järnbrist, medan ett högt värde är mindre specifikt för järnöverskott, eftersom en mängd andra orsaker kan orsaka hyperferritinemi [2-3]. Samtidig analys av transferrinmättnad är nödvändig för att bedöma om det finns ett järnöverskott. Den övre gränsen för normal transferrinmättnad anges vanligen till 0,45 (45 procent) för kvinnor och 0,50 (50 procent) för män, men den kan variera mellan olika klinisk-kemiska laboratorier. Denna översikt belyser hur hyperferritinemi utreds och när den bör behandlas.
Kroppens järnbalans
Ferritin är ett cytosoliskt protein som kan binda 4 500 järnjoner (Fe3+). Ferritinsyntesen ökar vid inflammation och vid tillstånd med stora järndepåer. Genom att ferritin sekvestrerar fritt järn och oxiderar Fe2+ till det mindre reaktiva Fe3+ så motverkas oxidativ stress, varför det kan betraktas som en antioxidant. Ferritin finns i stor mängd i leverceller och retikuloendoteliala celler (makrofager). Skador på dessa leder till läckage till blodbanan och mycket höga ferritinvärden i serum. Ferritin utsöndras också aktivt till serum i glykosylerad form, vilket ger en längre halveringstid och mer stabila serumnivåer som avspeglar kroppens järndepåer.
Järnmängden hos en vuxen är cirka 3–4 g. I genomsnitt finns 2,5 g bundet i hemoglobin i röda blodkroppar och erytroblaster, 400 mg i järnhaltiga proteiner, 3–7 mg i form av transferrinbundet järn och resten lagrat som ferritin eller hemosiderin. Kroppen förlorar i genomsnitt 1 mg järn per dag genom svettning eller förlust av hud- och tarmceller. Kvinnor förlorar mer än män till följd av menstruation och graviditet. I genomsnitt konsumerar en vuxen 1–2 mg hemjärn och 10–15 mg icke-hemjärn per dag, varav 30 procent respektive 10 procent absorberas, vilket motsvarar 1–2 mg järn per dag. Dessa genomsnittsnivåer varierar med kostens sammansättning, exempelvis förekomsten av polyfenoler, fytinsyra och kött. Järn absorberas i duodenum och proximala jejunum. Absorptionen av järn regleras av hormonet hepcidin [4], som bildas i levern och blockerar järnupptaget i tarmen och järnfrisättningen från makrofagerna [5]. Brist på hepcidin, vilket ses vid hereditär hemokromatos, leder till en högre järnabsorption i tarmen och en ökad järnfrisättning från makrofagerna, med en förhöjd transferrinmättnad i serum som resultat [6].
Initial bedömning av hyperferritinemi
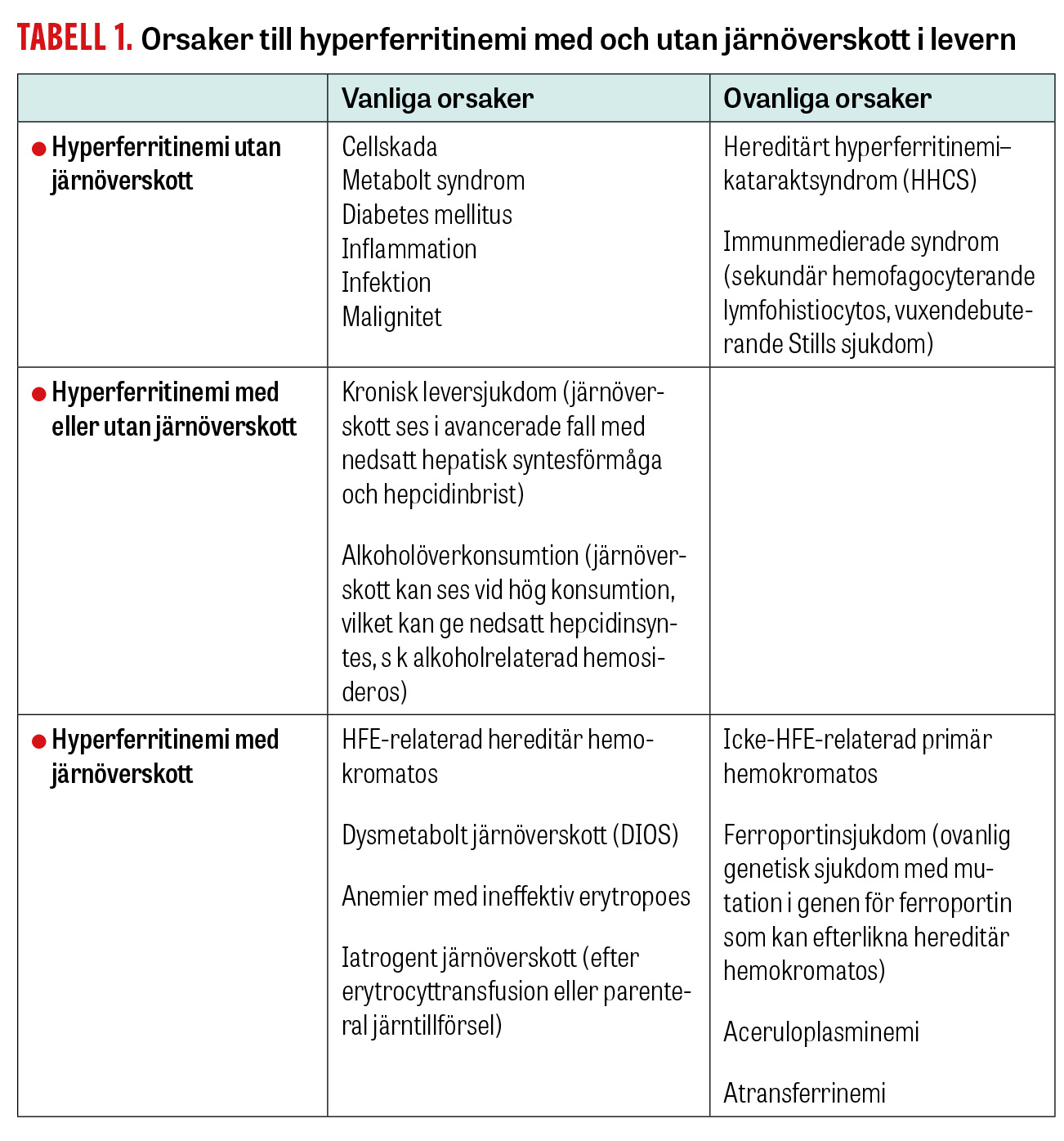
Markant förhöjt ferritin (>10 000 μg/l) bör föranleda övervägande av sällsynta tillstånd som hemofagocyterande lymfohistiocytos, vuxendebuterad Stills sjukdom [7] eller andra akuta tillstånd vid njursjukdom, leversjukdom, infektion eller malignitet [8]. I öppenvård ses vanligen en måttlig ferritinstegring (500–1 500 μg/l), som i de flesta fall orsakas av alkoholöverkonsumtion, kronisk inflammation eller metabolt syndrom. Vid hög transferrinmättnad bör man misstänka hereditär hemokromatos. Olika former av järnöverskott är orsak till högt ferritin i högst 10 procent av fallen; resten har annan genes. De vanligaste orsakerna till högt ferritin listas i Tabell 1.
Högt ferritin med normal transferrinmättnad – orsaker
Alkoholöverkonsumtion är en mycket vanlig orsak till högt ferritin, och mekanismen är en kombination av minskad hepcidinsyntes, läckage från skadade leverceller och kronisk inflammation. Alkoholkonsumtion >20 g per dag inducerar hyperferritinemi hos 40–70 procent [9, 10]. Vid mycket hög och långvarig alkoholkonsumtion kan transferrinmättnaden temporärt bli förhöjd och ferritin stiga till >1 000 μg/l, vilket kan simulera hemokromatos. I dessa fall ses vanligen en ASAT/ALAT-kvot >1 och fluktuerande ferritinnivå och transferrinmättnad. De kan utveckla en lindrig järninlagring i levern, så kallad hemosideros.
Vid MASLD (metabolic dysfunction-associated steatotic liver disease; tidigare NAFLD, non-alcoholic fatty liver disease), som är associerad till övervikt, metabolt syndrom och insulinresistens, ses förhöjt ferritin, så kallad dysmetabol hyperferritinemi, hos en tredjedel av patienterna [11]. Hos 20–30 procent av dessa ses även en lindrig järninlagring i levern, vilket benämns dysmetabolt järnöverskott (dysmetabolic iron overload syndrome, DIOS) [12-13]. Man anser att insulinresistens är en bakomliggande orsak. I hälften av fallen har patienten också MASLD [14]. Data tyder på att dysmetabolt järnöverskott ger en något ökad risk för diabetes mellitus, kardiovaskulär sjukdom och leversjukdom vid MASLD [15]. Dysmetabol hyperferritinemi har en normal transferrinmättnad, och även vid dysmetabolt järnöverskott är den oftast normal, eller endast lätt stegrad.
Är CRP eller SR förhöjt bör inflammation, infektion eller malignitet övervägas [7]. Vid autoimmuna sjukdomar och cancer ses hyperferritinemi på grund av såväl inflammation som cellulär skada [16, 17]. Måttlig förhöjning av ferritin ses hos 23 procent av patienterna med systemisk lupus erythematosus (SLE), men det kan också ses vid diabetes mellitus typ 1, multipel skleros och reumatoid artrit [18].
Blodmalignitet och solid cancer kan inducera förhöjda ferritinnivåer som resultat av inflammation och cytolys [16, 19]. Ferritinnivåer >1 000 μg/l kan ses vid metastaserande cancer [20].
Levern är det organ som lagrar mest ferritin. En akut hepatocellulär skada frisätter intracellulärt ferritin som kan nå plasmanivåer på >10 000 μg/l, vilket kan ses vid akut leversvikt eller paracetamolintoxikation. Ett kännetecken är att leverenzymer (ALAT, ASAT) är motsvarande kraftigt förhöjda [9, 21].
Vid sällsynta immunmedierade tillstånd som hemofagocyterande lymfohistiocytos producerar och frisätter monocyter och makrofager ferritin i höga nivåer, med serumvärden >10 000 μg/l [7]. Vid vuxendebuterande Stills sjukdom är ferritinvärden på 4 000–30 000 μg/l inte ovanliga [22–24].
Hereditärt hyperferritinemi–kataraktsyndrom (HHCS) är en sällsynt autosomalt dominant sjukdom som orsakas av en mutation i genen för L-ferritin. Mutationen leder till en okontrollerad produktion av järnfattigt L-ferritin, som deponeras i ögats lins och ger katarakt i unga år. Ferritinnivåerna ligger mellan 1 000 och 3 000 μg/l. Tillståndet är i övrigt ofarligt och leder inte till järnöverskott [25].
En annan sällsynt genetisk sjukdom relaterad till hyperferritinemi med normal transferrinmättnad är Gauchers sjukdom. Oförklarad cytopeni med hepatosplenomegali och högt ferritin kan väcka misstanke om sjukdomen [26-27].
Högt ferritin med järnöverskott – orsaker
Hög transferrinmättnad kan bero på högt serumjärn eller på lågt transferrin i serum [9]. Högt serumjärn ses vid primär och sekundär hemokromatos, medan lågt serumtransferrin kan bero på avancerad levercirros med nedsatt proteinsyntesförmåga i levern eller mycket ovanliga ärftliga tillstånd såsom atransferrinemi. Vid en mycket hög transferrinmättnad binds järnet även till andra plasmaproteiner (icke-transferrinbundet järn, NTBI) som lätt kan tas upp av parenkymatösa organ, främst levern.
Järnöverskott kan indelas i primärt, då orsaken är en ärftlig defekt i regleringen av järnbalansen, eller sekundärt som resultat av andra ärftliga eller förvärvade tillstånd [28]. Alkoholrelaterad hemosideros och dysmetabolt järnöverskott, vilka nämnts ovan, tillhör bägge gruppen sekundärt järnöverskott, som dock är lindrigt (hemosideros). Mer uttalad järninlagring, vilket benämns sekundär hemokromatos, kan ses efter upprepade blodtransfusioner eller parenteral järntillförsel. Typfallen är patienter med olika anemier med ineffektiv erytropoes (till exempel beta-talassemi eller dyserytropoetisk, aplastisk, sideroblastisk eller hemolytisk anemi) eller kronisk njursvikt med anemi. Efter kumulativ transfusion av >40 enheter erytrocytkoncentrat kan parenkymatöst järnöverskott utvecklas [29]. Histologiskt ses ansamling av järn i mononukleära fagocyter i retikulo-endoteliala systemet och i leverns makrofager (Kupfferceller) [30]. Anemier med ineffektiv erytropoes ger också en ökad järnabsorption, eftersom hepcidin nedregleras av både anemin och hypoxin [31-32].
Den vanligaste formen av primärt järnöverskott är HFE-associerad hereditär hemokromatos. HFE är ett cellyteprotein i levercellens membran som bildar komplex med transferrinreceptor 1 och 2 (TfR1 och TfR2) och proteinet hemojuvelin. Komplexet känner av järnmängden i plasma och signalerar till cellkärnan att öka syntesen av hepcidin vid höga järnnivåer. Hepcidin utsöndras av hepatocyterna och binder till »järnpumpen« ferroportin i cellmembranet på tarmepitelceller och retikuloendoteliala celler. Ferroportinets uppgift är att transportera ut järnjoner till plasma, där de binds till transferrin. Ferroportin degraderas när det binds till hepcidin. Vid hemokromatos orsakar det defekta HFE-proteinet en hepcidinbrist, varför ferroportinet har en fortsatt hög aktivitet och järnhalten i plasma förblir stegrad. Förutom mutation i HFE-genen kan även mutationer i generna för TfR2, hemojuvelin (HJV), hepcidin (HAMP) eller ferroportin (SLC40A1) orsaka hemokromatos. Dessa varianter är betydligt mer sällsynta än HFE-associerad hemokromatos.
Man har visat att cirka 80–90 procent av patienter med hereditär hemokromatos är homozygota för mutationen p.C282Y i HFE [33]. Denna mutation orsakar ett defekt HFE-protein som inte når cellytan. Mutationen uppstod i den keltiska folkstammen och har därför högst frekvens på Irland, där 11,7 procent av befolkningen bär på anlaget [34]. Därifrån har den spridit sig till Nordeuropa, USA och Australien. Den biokemiska penetransen för p.C282Y-homozygoter är endast 60–80 procent. Eftersom sjukdomen nedärvs autosomalt recessivt så får inte heterozygoter klinisk hereditär hemokromatos, även om deras järnparametrar kan ligga i övre delen av normalområdet. En annan HFE-mutation, p.H63D, är vanligare och ses över hela världen. Den påverkar HFE-proteinets affinitet till TfR2 men orsakar i sig inte hemokromatos. Även om H63D-homozygoter kan ha något förhöjt ferritin och transferrinmättnad, är det osannolikt att de utvecklar kliniskt järnöverskott [35-36]. Individer med blandad heterozygoti, det vill säga p.C282Y/H63D, kan utveckla hemokromatos, men risken är lägre och fenotypen lindrigare än hos p.C282Y-homozygoter [37]. Man bör därför leta efter ytterligare riskfaktorer vid denna genotyp.
De cirka 10 procent av hemokromatospatienterna som saknar HFE-mutationer har sannolikt mutationer i någon av de andra järnreglerande generna. Gensekvensering av TfR2, SLC40A1, HAMP och HJV kan göras på flera laboratorier i Sverige och kan övervägas i utvalda fall, speciellt på patienter med allvarlig fenotyp. I klinisk rutin brukar dessa dock sällan eftersökas, utan man behandlar vanligen på fenotypen för hereditär hemokromatos. Denna fenotyp inkluderar 1) en hög transferrinmättnad i serum, 2) ett högt serumferritin, och 3) en förhöjd järnmängd i levern. Vid p.C282Y homozygoti, eller blandad heterozygoti (det vill säga p.C282Y/H63D) räcker det att kriterierna 1 och 2 är uppfyllda, medan man vid andra HFE-genotyper eller normal HFE-genotyp även måste påvisa ett järnöverskott i levern. Detta görs med magnetresonanstomografi (MR) med speciella protokoll som kvantifierar järnmängden. Vid kontraindikation för MR kan järnöverskott påvisas med leverbiopsi.
Typiska symtom vid hereditär hemokromatos är trötthet, ledbesvär och i avancerade fall gråbrun missfärgning av huden. Män insjuknar oftare än kvinnor, och sjukdomsprevalensen ökar med åldern [38]. Obehandlad kan sjukdomen resultera i leverfibros, levercirros och hepatocellulär cancer [39-40]. Risken för levercirros anges till cirka 1–5 procent. Andra manifestationer inkluderar diabetes, osteoporos och artropati [41]. Artropatin drabbar främst småleder, som vrister, knogar och fingrar, men även knän. Kliniskt ter den sig som artros snarare än artrit. Svår eller tidigt debuterande hereditär hemokromatos, vilket är sällsynt, kan leda till hypogonadism, hypotyreos och hjärtsvikt [42].
En differentialdiagnos till hereditär hemokromatos är porphyria cutanea tarda, som orsakas av nedsatt aktivitet i enzymet uroporfyrinogendekarboxylas (UPGD) i levercellerna. Patienter med porphyria cutanea tarda har en något ökad förekomst av hemokromatos, varför HFE-gentestning rekommenderas [43].
Utredning av hyperferritinemi i primärvård
Anamnesen bör inkludera eventuell hereditet för hemokromatos och en uppskattning av antalet standardglas alkohol som konsumeras per vecka. Man frågar om samtidiga metabola (diabetes, hypertoni, hyperlipidemi), inflammatoriska, hematologiska, maligna eller renala sjukdomar. Finns det ledbesvär, speciellt i småleder? Har det funnits långvarig exponering för järnpreparat eller upprepade blodtransfusioner?
I status noteras eventuell övervikt eller fetma (BMI), blodtryck, tecken till levercirros (spider nevi, palmarerytem) samt tecken till ledpåverkan (uppdrivningar, värk och nedsatt rörlighet av småleder såsom fingrar, knogar och fotleder).
Laboratoriepanelen bör inkludera blodstatus, CRP, ASAT, ALAT, ALP, GT, PK(INR), bilirubin, fasteglukos och HbA1c, lipidprofil, fosfatidyletanol (PEth) samt ferritin och transferrinmättnad. Ferritin och transferrinmättnad bör undersökas vid minst 2 olika tillfällen eftersom värdena kan fluktuera.
Vid förhöjd transferrinmättnad (>0,45 för kvinnor och >0,50 för män) eller vid S-ferritin >1 000 μg/l rekommenderas mutationsanalys av HFE-genen för att fastställa eller utesluta hereditär hemokromatos. Huruvida HFE-analysen görs i primärvård eller senare i utredningen beror på lokala riktlinjer i respektive region.
Remiss till internmedicinare eller gastroenterolog rekommenderas vid förhöjd transferrinmättnad i minst 2 upprepade prov, vid S-ferritin >1 000 μg/l eller vid tecken till leverpåverkan eller misstanke om levercirros. Det betyder att patienter med ferritin <1 000 μg/l, normal transferrinmättnad och utan leverpåverkan eller cirrosmisstanke som regel kan handläggas inom primärvården.
Handläggning inom primärvården
Patienter med metabolt syndrom och hyperferritinemi har alltid en normal transferrinmättnad. De har ofta samtidigt fettlever (MASLD). Åtgärden är här viktminskning, alkoholfrihet och ökad motion. Ferritin följs årligen, och remiss till gastroenterolog skickas endast om ferritin stiger >1 000 μg/l. Vid MASLD bör man komplettera med analysverktyget FIB-4 för att utesluta avancerad leverfibros (se www.fib4.se).
Om patienten har en hög alkoholkonsumtion (>14 standardglas per vecka för män eller >9 för kvinnor) eller PEth >0,3 rekommenderas avhållsamhet från alkohol i 2–3 månader och därefter nytt prov. Remiss till internmedicinare eller gastroenterolog skickas endast om de höga ferritinvärdena kvarstår efter alkoholfrihet.
Om man uppdagar en kronisk inflammation, blodsjukdom, njursjukdom, eller malignitet handlägger man naturligtvis den underliggande sjukdomen, och inte ferritinvärdet i sig.
Varför ett gränsvärde för ferritin >1 000 µg/l ?
Gränsvärdet 1 000 μg/l bygger på två fakta:
- Vid hereditär hemokromatos har man visat att patienter med ferritin <1 000 μg/l inte utvecklar någon leverskada på grund av järnöverskottet.
- Vid dysmetabol hyperferritinemi har man visat att det är ovanligt med järninlagring i levern (DIOS) hos patienter med ferritin <1 000 μg/l.
Detta innebär att ett persisterande ferritin >1 000 μg/l efter alkoholfrihet i 2–3 månader bör utredas hos specialist, vanligen medicinsk gastroenterolog. Transferrinmättnad >0,45/0,50 bör också utredas hos specialist oavsett ferritinnivå, eftersom detta har hög sensitivitet för hereditär hemokromatos.
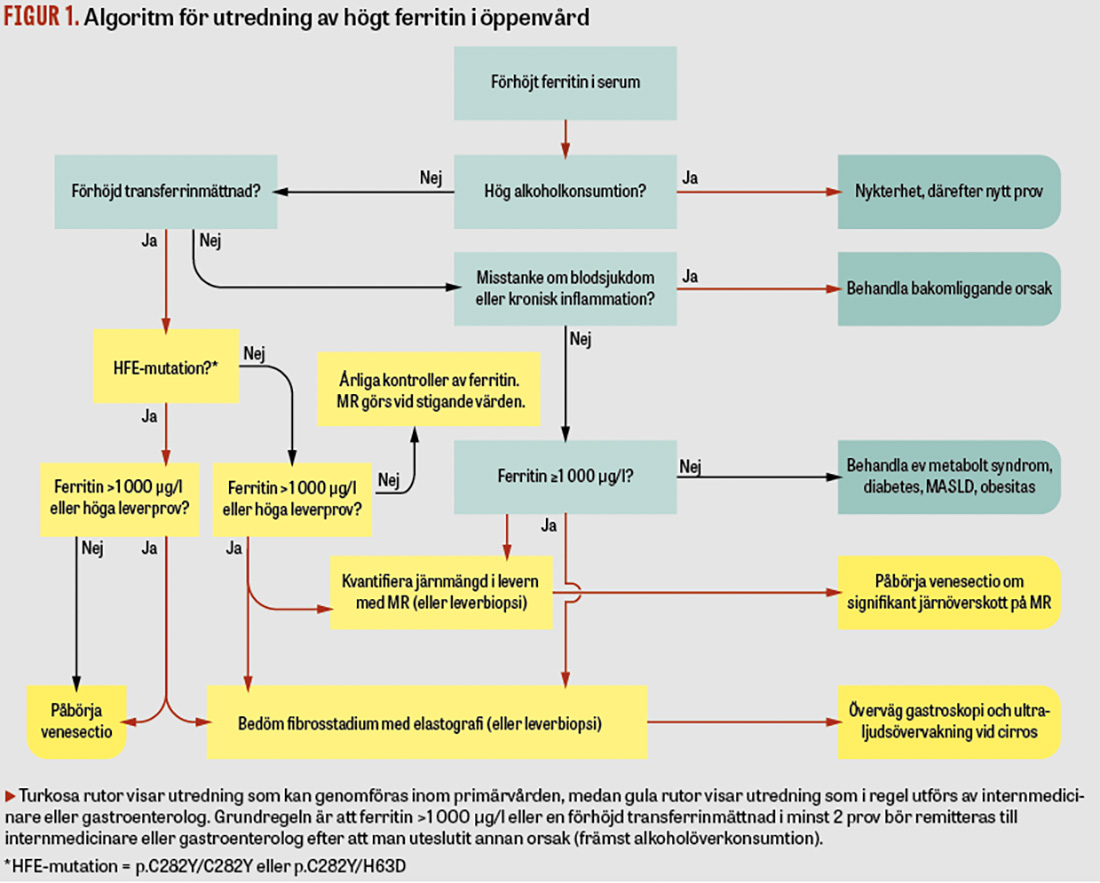
Olika algoritmer för utredning av högt ferritin har presenterats tidigare [44]. Vi föreslår här en algoritm för utredning av hyperferritinemi i öppenvård anpassad efter svenska förhållanden (se Figur 1).
Vidare handläggning (internmedicin/gastroenterologi)
Vid förhöjd transferrinmättnad och stegrat ferritin kontrolleras HFE-genen. Om denna visar homozygoti för p.C282Y eller blandad heterozygoti för p.C282Y/H63D påbörjas behandling med venesectio. Om HFE-analysen inte visar någon av dessa genotyper görs MR enligt hemokromatosprokoll. Om man då kan påvisa en ökad järnmängd i levern påbörjas behandling med venesectio. Vid kontraindikation för MR görs i stället leverbiopsi.
Om en patient med diagnosen hereditär hemokromatos har ett ferritinvärde >1 000 μg/l eller stegrade transaminaser rekommenderas elastografi för att bedöma om det finns tecken till cirros eller avancerad fibros. Om elastografin visar cirros rekommenderas ultraljudövervakning, eftersom det föreligger en hög risk för hepatocellulär cancer. Vid svårtolkade elastografivärden kan man överväga leverbiopsi.
Patienter med normal transferrinmättnad men ferritin >1 000 μg/l har inte fenotypen för hereditär hemokromatos, men kan ha en viss järninlagring i levern på grund av dysmetabolt järnöverskott eller alkoholrelaterad hemosideros. MR enligt hemokromatosprotokoll rekommenderas i dessa fall för att kvantifiera mängden järn. Det råder inte konsensus om huruvida dessa patienter bör behandlas med venesectio, eftersom deras järnöverskott ofta är lindrigt [45]. Man anser dock att en nivå över 7 mg järn/g lever (motsvarande 125 µmol/g) ger en något ökad risk för järninducerad leverpåverkan, och en nivå >15 mg/g (>269 µmol/g) ger en tydligt ökad risk [46]. I andra riktlinjer, såsom UpToDate, rekommenderas venesectio vid värden över 4–5 mg järn/g lever. Man kan därför överväga venesectio vid dysmetabolt järnöverskott eller alkoholrelaterad hemosideros om utredning med MR visar värden över den lägre av dessa nivåer och rekommendera venesectio vid den högre nivån.
Behandling
Vid diagnostiserad hereditär hemokromatos görs venesectio varje till varannan vecka till målferritinvärde 50–100 μg/l. Detta riktmärke kan anpassas uppåt för äldre eller sköra patienter. Vanligen tappar man 450 ml vid varje tillfälle, men vid låg kroppsvikt eller hos äldre patienter kan mängden blod eller tappningsintervallet behöva individanpassas. Hemoglobin kontrolleras vid varje tappningstillfälle. Om Hb är <120 g/l minskas tappningsmängden till 300 ml, och vid Hb <110 g/l görs uppehåll med tappning. Kobalamin och folat i serum bör kontrolleras vid lågt Hb. När man uppnått målet avslutas intensivbehandlingen och man övergår till underhållsbehandling med venesectio 2–6 gånger per år. S-ferritin kontrolleras årligen, och nästkommande års tappningsintervall justeras efter målferritin <100 μg/l. I övrigt friska patienter med hereditär hemokromatos kan bli blodgivare under underhållsfasen. Ferritin bör kontrolleras årligen i dessa fall.
Vid behandling av dysmetabolt järnöverskott eller alkoholrelaterad hemosideros med höga järnmängder i levern (se gränsvärden ovan) görs venesectio med något glesare intervall än vid hemokromatos, exempelvis en tappning per månad. Målferritinvärde kan också ligga högre (<300 μg/l) eftersom ferritinvärdet generellt är högre vid motsvarande järninlagring jämfört med hereditär hemokromatos. Ferritinvärdet bör kontrolleras årligen. Underhållsbehandling är vanligen inte nödvändig.
Screening
Syskon till en patient med hereditär hemokromatos screenas för sjukdomen med kontroll av transferrinmättnad och S-ferritin, och om patienten har en HFE-mutation bör även HFE-analys göras. Barn screenas på samma sätt efter 18 års ålder. Om hereditär hemokromatos upptäcks och behandlas i tidigt skede kan man förebygga komplikationer och få en normal livslängd.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Referenser
- Ogilvie C, Fitzsimons K, Fitzsimons EJ. Serum ferritin values in primary care: are high values overlooked? J Clin Pathol. 2010;63(12):1124-6.
- Zanella A, Gridelli L, Berzuini A, et al. Sensitivity and predictive value of serum ferritin and free erythrocyte protoporphyrin for iron deficiency. J Lab Clin Med. 1989;113(1):73-8.
- Knovich MA, Storey JA, Coffman LG, et al. Ferritin for the clinician. Blood Rev. 2009;23(3):95-104.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102(3):783-8.
- Détivaud L, Nemeth E, Boudjema K, et al. Hepcidin levels in humans are correlated with hepatic iron stores, hemoglobin levels, and hepatic function. Blood. 2005;106(2):746-8.
- Pietrangelo A. Hereditary hemochromatosis – a new look at an old disease. N Engl J Med. 2004;350(23):2383-97.
- Rosário C, Zandman-Goddard G, Meyron-Holtz EG, et al. The hyperferritinemic syndrome: macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013;11:185.
- Schram AM, Campigotto F, Mullally A, et al. Marked hyperferritinemia does not predict for HLH in the adult population. Blood. 2015;125(10):1548-52.
- Adams PC, Barton JC. A diagnostic approach to hyperferritinemia with a non-elevated transferrin saturation. J Hepatol. 2011;55(2):453-8.
- Moirand R, Lescoat G, Delamaire D, et al. Increase in glycosylated and nonglycosylated serum ferritin in chronic alcoholism and their evolution during alcohol withdrawal. Alcohol Clin Exp Res. 1991;15(6):963-9.
- Ioannou GN, Weiss NS, Kowdley KV. Relationship between transferrin-iron saturation, alcohol consumption, and the incidence of cirrhosis and liver cancer. Clin Gastroenterol Hepatol. 2007;5(5):624-9.
- Riva A, Trombini P, Mariani R, et al. Revaluation of clinical and histological criteria for diagnosis of dysmetabolic iron overload syndrome. World J Gastroenterol. 2008;14(30):4745-52.
- Moirand R, Mortaji AM, Loréal O, et al. A new syndrome of liver iron overload with normal transferrin saturation. Lancet. 1997;349(9045):95-7.
- Branisso PPF, de Oliveira CPMS, Filho HML, et al. Non-invasive methods for iron overload evaluation in dysmetabolic patients. Ann Hepatol. 2022;27(4):100707.
- Datz C, Müller E, Aigner E. Iron overload and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017;42(2):173-83.
- Wang W, Knovich MA, Coffman LG, et al. Serum ferritin: past, present and future. Biochim Biophys Acta. 2010;1800(8):760-9.
- Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99(10):3505-16.
- Orbach H, Zandman-Goddard G, Amital H, et al. Novel biomarkers in autoimmune diseases: prolactin, ferritin, vitamin D, and TPA levels in autoimmune diseases. Ann N Y Acad Sci. 2007;1109:385-400.
- Cullis JO, Fitzsimons EJ, Griffiths WJ, et al; British Society for Haematology. Investigation and management of a raised serum ferritin. Br J Haematol. 2018;181(3):331-40.
- Lorcerie B, Audia S, Samson M, et al. Diagnosis of hyperferritinemia in routine clinical practice. Presse Med. 2017;46(12 Pt 2):e329-38.
- Kell DB, Pretorius E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics. 2014;6(4):748-73.
- Fautrel B. Ferritin levels in adult Still’s disease: any sugar? Joint Bone Spine. 2002;69(4):355-7.
- Coffernils M, Soupart A, Pradier O, et al. Hyperferritinemia in adult onset Still’s disease and the hemophagocytic syndrome. J Rheumatol. 1992;19(9):1425-7.
- Ohta A, Yamaguchi M, Kaneoka H, et al. Adult Still’s disease: review of 228 cases from the literature. J Rheumatol. 1987;14(6):1139-46.
- Vaktnäs J, Olsson M. Hereditärt hyperferritinemi–kataraktsyndrom. Ovanlig differentialdiagnos vid oförklarlig ferritinstegring. Läkartidningen. 2013;110(51–52):2302-3.
- Mistry PK, Sadan S, Yang R, et al. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82(8):697-701.
- Nagral A. Gaucher disease. J Clin Exp Hepatol. 2014;4(1):37-50.
- Piperno A. Classification and diagnosis of iron overload. Haematologica. 1998;83(5):447-55.
- Kohgo Y, Ikuta K, Ohtake T, et al. Body iron metabolism and pathophysiology of iron overload. Int J Hematol. 2008;88(1):7-15.
- Siah CW, Ombiga J, Adams LA, et al. Normal iron metabolism and the pathophysiology of iron overload disorders. Clin Biochem Rev. 2006;27(1):5-16.
- Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366(4):348-59.
- Shenoy N, Vallumsetla N, Rachmilewitz E, et al. Impact of iron overload and potential benefit from iron chelation in low-risk myelodysplastic syndrome. Blood. 2014;124(6):873-81.
- Porto G, Brissot P, Swinkels DW, et al. EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet. 2016;24(4):479-95.
- European Association for the Study of the Liver. EASL clinical practice guidelines on haemochromatosis. J Hepatol. 2022;77(2):479-502.
- Gurrin LC, Bertalli NA, Dalton GW, et al; HealthIron Study Investigators. HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity. Hepatology. 2009;50(1):94-101.
- Gochee PA, Powell LW, Cullen DJ, et al. A population-based study of the biochemical and clinical expression of the H63D hemochromatosis mutation. Gastroenterology. 2002;122(3):646-51.
- Allen K. Hereditary haemochromatosis – diagnosis and management. Aust Fam Physician. 2010;39(12):938-41.
- Hagström H, Ndegwa N, Jalmeus M, et al; Swedish Hepatology Study Group (SweHep). Morbidity, risk of cancer and mortality in 3645 HFE mutations carriers. Liver Int. 2021;41(3):545-53.
- Atkins JL, Pilling LC, Masoli JAH, et al. Association of hemochromatosis HFE p.C282Y homozygosity with hepatic malignancy. JAMA. 2020;324(20):2048-57.
- Pilling LC, Tamosauskaite J, Jones G, et al. Common conditions associated with hereditary haemochromatosis genetic variants: cohort study in UK Biobank. BMJ. 2019;364:k5222.
- Sahinbegovic E, Dallos T, Aigner E, et al. Musculoskeletal disease burden of hereditary hemochromatosis. Arthritis Rheum. 2010;62(12):3792-8.
- Kelly AL, Rhodes DA, Roland JM, et al. Hereditary juvenile haemochromatosis: a genetically heterogeneous life-threatening iron storage disease. QJM. 1998;91(9):607-18.
- Stölzel U, Doss MO, Schuppan D. Clinical guide and update on porphyrias. Gastroenterology. 2019;157(2):365-81.e4.
- Sandnes M, Ulvik RJ, Vorland M, et al. Hyperferritinemia – a clinical overview. J Clin Med. 2021;10(9):2008.
- Adams LA, Crawford DH, Stuart K, et al. The impact of phlebotomy in nonalcoholic fatty liver disease: a prospective, randomized, controlled trial. Hepatology. 2015;61(5):1555-64.
- Reeder SB, Yokoo T, França M, et al. Quantification of liver iron overload with MRI: review and guidelines from the ESGAR and SAR. Radiology. 2023;307(1):e221856.
Summary
Ferritin is one of the most requested blood tests in both primary and inpatient care, and high values occur frequently. One of the greatest challenges in the investigation of hyperferritinemia is to determine if there is a presence of iron overload. Patient history (chronic liver disease, excessive alcohol consumption, hereditary factors), clinical features (metabolic syndrome, acute or chronic inflammation, infection, malignancy) and biochemical tests (ferritin, transferrin saturation, hemoglobin, liver enzymes, CRP/SR, phosphatidyl ethanol, lipid profile, glucose) facilitate the determination of the cause of hyperferritinemia. High transferrin saturation indicates iron overload, which is usually linked to hereditary hemochromatosis. In the case of homozygosity of the HFE mutation p.C282Y in the presence of hyperferritinemia, venesection can be started without further investigations, while in the absence of HFE mutations a possible iron excess must be validated with magnetic resonance imaging (MRI) for iron determination before venesection is started. Dysmetabolic iron overload syndrome (DIOS) or alcohol-related hemosiderosis can be treated with venesection in selected cases if there is a significant deposition of iron in the liver on MRI. An individual with ferritin below 1000 µg/L, a normal transferrin saturation, and normal liver tests does not need further investigations and can be followed in primary care. We propose an algorithm for the investigation of hyperferritinemia that facilitates the investigation both in primary and inpatient care.