Ovarialcancer betraktas i dag som ett samlingsbegrepp för ett antal sinsemellan molekylärbiologiskt helt separata tumörformer, med olika klinisk bild och prognos.
Ärftlig predisposition är den största riskfaktorn för ovarialcancer.
Mutationer i BRCA- och MMR-generna är de viktigaste ärftliga faktorerna.
Paritet, amning, hormonbehandling och vissa gynekologiska tillstånd påverkar risken att insjukna.
Man har hittills inte kunnat påvisa någon avgörande betydelse av livsstilsfaktorer för risken att utveckla ovarialcancer.
Såväl salpingooforektomi som salpingektomi sänker kraftigt risken att drabbas av ovarialcancer.
Epitelial ovarialcancer består av fem sjukdomar med olika ursprung och molekylärbiologisk, histologisk och klinisk bild: höggradig serös (70 procent), låggradig serös (<5 procent), endometrioid (10 procent), klarcellig (10 procent) och mucinös ovarialcancer (3–4 procent) [1, 2]. Dessutom finns en liten grupp tumörer som inte kan klassificeras närmare (ca 1 procent). Ingen av dessa celltyper har någon benign motsvarighet i det normala ovariet.
Patogenes
Patogenetisk forskning har varit svår att genomföra på grund av ovariets otillgänglighet. Adnexa biopseras vanligtvis inte och opereras bort endast på grund av patologiska processer.
Höggradig serös ovarialcancer (HGSC). Undersökning av salpingooforektomipreparat som opererats bort i riskreducerande syfte hos kvinnor med ärftlig bröst- och ovarialcancer (HBOC, hereditary breast and ovarian cancer) (se ärftlighet nedan, BRCA1- eller BRCA2-genmutationsbärare) har visat växt av serös tubar in situ-cancer (STIC), ett förstadium till invasion, i den distala fimbrieförsedda delen av tuban. Serös tubar in situ-cancer uppvisar sekretorisk cellmorfologi, höggradig kärnatypi, pleomorfism och hög mitosfrekvens [2].
Sekretoriska markörer, såsom mucin 1 och stathmin 1, är positiva [3]. Serös tubar in situ-cancer uppvisar dessutom mutationer i p53-genen och förekomst av DNA-skada [4]. Även tidigare lesioner har identifierats, vilket stödjer modellen för en stegvis förändring av den sekretoriska cellen [5]. Eftersom serös in situ-cancer utvecklas på tubans fimbrieyta, kan celler lossna och sprida sig över till det intilliggande ovariets yta och till peritoneum och oment.
Tillväxten av varje cellkluster är beroende av exakt vilka mutationer och kromosomavvikelser just den klonen har och kan leda till en blandad klinisk bild, där den största tumörmassan kan finnas på t ex ovarium, tuba eller peritoneum. P-piller, som skyddar mot ovarialcancer, hämmar ovulation, vilket alltså leder till färre epitelskador på ovariets yta. När dessa ska läka kan celler från serös in situ-cancer inkorporeras i ovariet eller på dess yta och senare utvecklas till cancer.
Huruvida denna modell, som har utvecklats vid studier av adnexa från mutationsbärande kvinnor, är allmänt applicerbar är oklart. Sporadiska (icke-ärftliga) cancerfall uppvisar oftast stora skrymmande tumörer på flera ställen, vilket hindrar möjligheten att identifiera prekursorer. Flera observationer stödjer dock denna modell. Hos 40–70 procent av kvinnorna med sporadisk höggradig serös ovarialcancer visar tuban antingen serös in situ-cancer eller invasiv cancer. Även om platsen för den initiala in situ-cancerutvecklingen kanske inte i samtliga fall är tuban, kan endosalpingios (ektopiskt benignt tubarepitel) påträffas i hela den kvinnliga bukhinnan. Sådana fokus kan genomgå en likadan cancerutveckling, även om lokalisationen inte är tuban [6].
Låggradig serös ovarialcancer (LGSC). Låggradig serös ovarialcancer har först nyligen erkänts som en egen sjukdomsentitet. Detta är också en malign tumör av serösa celler i tuban, men med ett helt annat ursprung, biologi och klinisk bild [2]. Låggradig serös ovarialcancer utvecklas från borderlinetumörer genom en definierad progression, där en del av de serösa cystadenomen utvecklas via serösa borderlinetumörer till låggradig serös cancer. Progressionen sker genom att cellerna förvärvat antingen KRAS- eller BRAF-mutationer.
Endometrioid och klarcellig ovarialcancer. Både endometrioid och klarcellig ovarialcancer kan utvecklas från endometrios, och ofta ses tumörnära atypisk endometrios. Samma molekylära defekter ses i karcinomen och i den atypiska endometriosen. Mutationer i PTEN i ARID1A verkar vara ett gemensamt steg för utveckling av endometrioid ovarialcancer [7, 8]. Förlust av BAF250, ett protein som spelar en central roll i kromatinmoduleringen, förekommer också. Mutationer i CTNNB1 (β-katenin) och PTEN är de mest frekventa mutationerna vid endometrioid ovarialcancer.
Klarcellig epitelial ovarialcancer är en av de ovarialtumörer som det råder mest oklarhet kring, eftersom det inte finns något motsvarande godartat »klarcelligt« epitel. Även denna tumörform ses ofta i samband med endometrios, vilket skulle kunna ge stöd för ett ursprung i endometriet [2]. Molekylära studier har identifierat mutationer i ARID1A i ungefär hälften av de klarcelliga tumörerna, som dessutom (även de) saknar uttryck av BAF250-proteinet [8].
Mucinös ovarialcancer. Ursprunget till mucinös epitelial ovarialcancer är oklart, men denna cancerform förmodas uppkomma genom utveckling från mucinösa borderlinetumörer. Bilden kompliceras av svårigheter att skilja mellan metastaser från gastrointestinalkanalen och ovarialcancer. Tumörepitelet kan uppvisa en histologi som liknar såväl endocervix som tjocktarms- och ventrikelepitel. Mutationer i KRAS ses i ca 80 procent av mucinösa borderlinetumörer och i 44 procent av mucinös epitelial ovarialcancer. Amplifiering av HER2 ses hos ca 6 procent av mucinösa borderlinetumörer och hos ca 18 procent av mucinös ovarialcancer. Sjukdomsprognosen är bättre om KRAS-mutationer eller HER2-amplifiering föreligger [9].
Etiologi
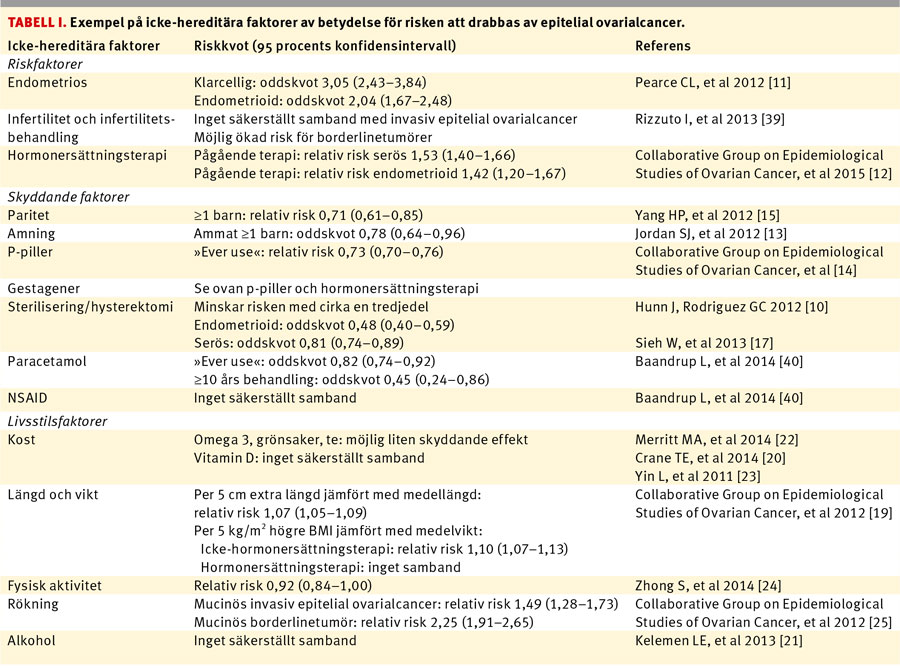
Riskfaktorer. Ärftlighet är den enskilt största riskfaktorn för ovarialcancer, men reproduktiv anamnes, vissa gynekologiska tillstånd och livsstilsfaktorer påverkar också risken att drabbas. Tabell I sammanfattar ett antal icke-hereditära faktorer. Den risk alternativt det skydd dessa faktorer medför skiljer sig mellan de olika subtyperna av ovarialcancer.
Endometrios dubblerar risken för endometrioid ovarialcancer och tredubblar risken för den klarcelliga subtypen, men synes inte ge upphov till den vanligast förekommande höggradigt serösa cancern [10]. Infertilitet och infertilitetsbehandling har länge ansetts öka risken för äggstockscancer, men en nylig Cochraneanalys har inte kunnat fastslå något samband mellan ofrivillig barnlöshet och invasiv ovarialcancer [11].
Substitutionsterapi med östrogen i klimakteriet, med eller utan tillägg av gestagen, medför en måttlig ökning av risken för ovarialcancer [12]. Denna risk avtar efter utsättning, men ännu efter 10 år ses ökad risk för serösa och endometrioida tumörer.
Skyddande faktorer. En fullgången graviditet minskar risken för ovarialcancer med upp till en tredjedel, skyddet ökar med antal barnafödslar [10]. Även amning har skyddande effekt [13].
Det skydd p-piller ger ökar med behandlingstiden. Riskminskningen är ca 6 procent per år och kvarstår efter utsättning i över 30 år (om än försvagat med tiden) [14]. Den skyddande effekten av p-piller verkar begränsad till den höggradigt serösa undergruppen [15]. P-piller kan användas profylaktiskt för BRCA-mutationsbärare med åtföljande halverad risk för ovarialcancer [8]. Gestagenbaserade p-piller och s k minipiller skyddar också [16].
Sterilisering och/eller hysterektomi minskar risken att insjukna med cirka en tredjedel [10, 17, 18].
Livsstilsfaktorer. Metaanalyser rörande betydelsen av kostens sammansättning, kroppslängd, vikt, BMI, fysisk aktivitet och alkoholbruk för risken att drabbas har visat endast ringa påverkan av dessa livsstilsfaktorer [19-24]. Rökning ökar dock signifikant risken för mucinösa tumörer [25].
Ärftlighet
Om en kvinna har en förstagradssläkting med ovarialcancer, har hon 2–6 gånger så stor risk att drabbas som andra kvinnor. Detta talar för en genetisk koppling [12]. Ärftlig ovarialcancer kan huvudsakligen delas in i ärftlig bröst- och ovarialcancer (HBOC) och Lynchs syndrom (kallas även ärftlig icke-polypös kolorektal cancer, HNPCC). Ärftlig bröst- och ovarialcancer dominerar. Andelen ärftlig ovarialcancer är större än man tidigare trott. Ca 20–25 procent av all serös epitelial ovarialcancer är ärftlig [26, 27]. Låg insjuknandeålder, synkron bröst- och ovarialcancer och familjehistoria (autosomalt dominant nedärvningsmönster) talar för ärftlig genes.
Ärftlig bröst- och ovarialcancer. Ärftlig bröst- och ovarialcancer står för ca 80 procent av all ärftlig ovarialcancer och är framför allt förknippad med BRCA (breast cancer susceptibility genes)-mutationer. På senare tid har även andra gener kopplats till ärftlig bröst- och ovarialcancer, framför allt gener längs BRCA-signaleringsvägen [28] (se nedan om mutationsscreening).
Livstidsrisken för att drabbas av ovarialcancer hos en mutatonsbärare är för BRCA1-bärare ca 40 procent och för BRCA2-bärare 10 procent. Det bör tilläggas att risken att insjukna i bröstcancer också är kraftigt förhöjd, ca 65 procent och 45 procent för respektive gen [29]. Detta ska jämföras med livstidrisken för ovarialcancer hos populationen i Sverige, ca 1,1 procent [30].
Cirka 65–75 procent av mutationsbärarna har familjehistoria [31]. BRCA1- och BRCA2-bärarfenotyperna är snarlika varandra. Möjligen har BRCA2-bärarna något högre insjuknandeålder (55–58 år) än BRCA1-bärarna (49–53 år) [26]. Insjuknandeåldern för bröstcancer är i regel lägre. Den vanligaste tumörhistologin är höggradigt seröst adenokarcinom. Lågt differentierat adenokarcinom av endometrioid typ kan förekomma och, mer sällan, klarcellig histologi. Mucinöst adenokarcinom är också ovanligt [32].
Ovarialcancer hos en BRCA-bärare svarar ofta bra på platinabaserad kemoterapi. Generellt, om optimal onkologisk behandling har kunnat ges, är prognosen något bättre för mutationsbärare än för patienter med sporadiska tumörer [33].
Lynchs syndrom. Lynchs syndrom är den näst vanligaste hereditära orsaken till ovarialcancer och står för ca 10–15 procent av all ärftlig ovarialcancer (vilket innebär mindre än 1 procent av all ovarialcancer) [20]. Dessa patienter bär på mutationer i »mismatch repair« (MMR)-generna. Mutationerna medför mikrosatellitinstabilitet i DNA-molekylen, vilket i sin tur leder till påverkan på apoptos och cellsignalering. Mutationsbärarna har kraftigt ökad risk att drabbas av framför allt koloncancer (livstidsrisk ca 50 procent) och endometriecancer (livstidsrisk 40–60 procent), men även ovarialcancer.
Livstidsrisken för ovarialcancer är 8–12 procent. De flesta insjuknar mellan 42 och 49 års ålder. Histologin är varierande; endometrioid ovarialcancer är emellertid vanligare även om alla subtyper förekommer [2]. Prognosen för mutationsbärare med ovarialcancer är bättre än för kvinnor med ärftlig bröst- och ovarialcancer eller sporadisk ovarialcancer [34].
Riskminskning för mutationsbärare. Det finns för närvarande inget bevis för att populationsscreening skulle upptäcka ovarialcancer tidigare. Data talar däremot för att mutationsbärare kraftigt reducerar sin risk att drabbas av ovarialcancer om en profylaktisk bilateral salpingooforektomi (med eller utan hysterektomi) genomförs i 40–45 års ålder eller när barnafödandet är avslutat. Flertalet gjorda studier indikerar en risksänkning på upp till 80 procent [35].
I en nyligen presenterad svensk populationsbaserad kohortstudie har man kunnat visa att även bilateral salpingektomi väsentligen sänker risken att drabbas av ovarialcancer. Detta antyder att kvinnor som tillhör riskpopulationen inte skulle behöva ooforektomeras i förtid [36]. P-piller har också visat sig skydda mot ovarialcancer, hos både BRCA-mutationsbärare och icke-mutationsbärare [37].
Mutationsscreening
BRCA1- och BRCA2-generna kodar för multifunktionella proteiner, som främst kan kopplas till reparation av dubbelsträngade DNA-skador via homolog rekombination. Patienter med nedärvd mutation i den ena kopian av BRCA1 eller BRCA2 har uttalad predisposition för bröst- och ovarialcancer. De bär redan från födseln en mutation i kroppens alla celler. När även den kvarvarande normala kopian av BRCA-genen inaktiveras (en andra punktmutation, deletion eller promotormetylering) i en somatisk cell förloras en essentiell funktion, vilket leder till ansamling av DNA-skador och risk för cancerutveckling.
De flesta BRCA-mutationer är nedärvda och har ett populationsspecifikt mönster. I Sverige ses en överrepresentation av vissa BRCA1-mutationer, s k foundermutationer (en ärftlig mutation som funnits i delar av befolkningen en längre tid), som nått en större spridning i befolkningen och som påträffas i till synes oberoende familjer. Dock kan frekvensen nymutationer vara underskattad, eftersom testning hittills huvudsakligen utgått från familjär bröst- och ovarialcancer. Senare studier visar att 30 procent av ovarialcancerpatienterna med ärftlig BRCA-mutation saknar familjär historia för sjukdomarna [28].
Metoder för mutationsscreening. Screening för BRCA-mutationer sker numera med modern teknik: NGS (nya generationens sekvensering) av de anrikade (amplikon eller »hybrid capture«) genomregionerna. Med bioinformatik kan sjukdomsassocierade sekvensvarianter som orsakar prematura stoppkodoner, känsliga aminosyrautbyten, felaktig splitsning eller större deletioner/duplikationer detekteras och urskiljas från normala sekvensvarianter.
Inom cancergenetiken söker man traditionellt efter nedärvda mutationer och utgår då från DNA extraherat från blod från indexpatient. Även somatiska mutationer kan dock bli aktuella i behandlingsprediktivt syfte. Samma NGS-metoder kan appliceras även på färsk/frusen tumörvävnad, med hänsyn tagen till att muterad/normal gen då kan finnas i annat förhållande än det heterozygota 50:50 som förväntas i blodcellerna. Även DNA från formalinfixerad vävnad går att använda men är av lägre kvalitet, kräver annan form av sekvensering och begränsar möjligheten till heltäckande analys.
NGS ger annars en potential att analysera betydligt fler gener än BRCA1 och BRCA2, och screening av genpaneler används både inom diagnostik och i forskningssyfte för att ge en mer nyanserad bild av patientens risk- och/eller tumörprofil [28, 38].
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Referenser
- Gilks CB, Prat J. Ovarian carcinoma pathology and genetics: recent advances. Hum Pathol. 2009;40:1213-23.
- Prat J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012;460:237-49.
- Karst AM, Levanon K, Duraisamy S, et al. Stathmin 1, a marker of PI3K pathway activation and regulator of microtubule dynamics, is expressed in early pelvic serous carcinomas. Gynecol Oncol. 2011;123:5-12.
- Carlson J, Roh MH, Chang MC, et al. Recent advances in the understanding of the pathogenesis of serous carcinoma: the concept of low- and high-grade disease and the role of the fallopian tube. Diagn Histopathol (Oxf). 2008;14:352-65.
- Jarboe E, Folkins A, Nucci MR, et al. Serous carcinogenesis in the fallopian tube: a descriptive classification. Int J Gynecol Pathol. 2008;27:1-9.
- Salvador S, Gilks B, Kobel M, et al. The fallopian tube: primary site of most pelvic high-grade serous carcinomas. Int J Gynecol Cancer. 2009;19:58-64.
- Sato N, Tsunoda H, Nishida M, et al. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 2000;60:7052-6.
- Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532-43.
- Anglesio MS, Kommoss S, Tolcher MC, et al. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. J Pathol. 2013;229:111-20.
- Hunn J, Rodriguez GC. Ovarian cancer: etiology, risk factors, and epidemiology. Clin Obstet Gynecol. 2012;55:3-23.
- Pearce CL, Templeman C, Rossing MA, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol. 2012;13:385-94.
- Collaborative Group on Epidemiological Studies of Ovarian Cancer; Beral V, Gaitskell K, Hermon C, et al. Menopausal hormone use and ovarian cancer risk: individual participant meta-analysis of 52 epidemiological studies. Lancet. 2015;385(9980):1835-42.
- Jordan SJ, Cushing-Haugen KL, Wicklund KG, et al. Breast-feeding and risk of epithelial ovarian cancer. Cancer Causes Control. 2012;23:919-27.
- Collaborative Group on Epidemiological Studies of Ovarian Cancer; Beral V, Doll R, Hermon C, et al. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet. 2008;371:303-14.
- Yang HP, Trabert B, Murphy MA, et al. Ovarian cancer risk factors by histologic subtypes in the NIH-AARP Diet and Health Study. Int J Cancer. 2012;131:938-48.
- Schuler S, Ponnath M, Engel J, et al. Ovarian epithelial tumors and reproductive factors: a systematic review. Arch Gynecol Obstet. 2013;287:1187-204.
- Sieh W, Salvador S, McGuire V, et al. Tubal ligation and risk of ovarian cancer subtypes: a pooled analysis of case-control studies. Int J Epidemiol. 2013;42:579-89.
- Sueblinvong T, Carney ME. Current understanding of risk factors for ovarian cancer. Curr Treat Options Oncol. 2009;10:67-81.
- Collaborative Group on Epidemiological Studies of Ovarian Cancer. Ovarian cancer and body size: individual participant meta-analysis including 25,157 women with ovarian cancer from 47 epidemiological studies. PLoS Med. 2012;9:e1001200.
- Crane TE, Khulpateea BR, Alberts DS, et al. Dietary intake and ovarian cancer risk: a systematic review. Cancer Epidemiol Biomarkers Prev. 2014;23:255-73.
- Kelemen LE, Bandera EV, Terry KL, et al. Recent alcohol consumption and risk of incident ovarian carcinoma: a pooled analysis of 5,342 cases and 10,358 controls from the Ovarian Cancer Association Consortium. BMC Cancer. 2013;13:28.
- Merritt MA, Cramer DW, Missmer SA, et al. Dietary fat intake and risk of epithelial ovarian cancer by tumour histology. Br J Cancer. 2014;110:1392-401.
- Yin L, Grandi N, Raum E, et al. Meta-analysis: circulating vitamin D and ovarian cancer risk. Gynecol Oncol. 2011;121:369-75.
- Zhong S, Chen L, Lv M, et al. Non-occupational physical activity and risk of ovarian cancer: a meta–analysis. Tumour Biol. 2014;35:11065-73.
- Collaborative Group on Epidemiological Studies of Ovarian Cancer; Beral V, Gaitskell K, Hermon C, et al. Ovarian cancer and smoking: individual participant meta-analysis including 28,114 women with ovarian cancer from 51 epidemiological studies. Lancet Oncol. 2012;13:946-56.
- Weissman SM, Weiss SM, Newlin AC. Genetic testing by cancer site: ovary. Cancer J. 2012;18:320-7.
- Mutch D, Denny L, Quinn M, et al. Hereditary gynecologic cancers. Int J Gynaecol Obstet. 2014;124:189-92.
- Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:18032-7.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117-30.
- Cancerincidens i Sverige 2013. Nya diagnostiserade cancerfall år 2013. Stockholm: Socialstyrelsen; 2014. Artikelnr 2014-12-10.
- Pal T, Permuth-Wey J, Betts JA, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104:2807-16.
- Brown J, Frumovitz M. Mucinous tumors of the ovary: current thoughts on diagnosis and management. Curr Oncol Rep. 2014;16:389.
- Alsop K, Fereday S, Meldrum C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30:2654-63.
- Nakamura K, Banno K, Yanokura M, et al. Features of ovarian cancer in Lynch syndrome (review). Mol Clin Oncol. 2014;2:909-16.
- Long KC, Kauff ND. Hereditary ovarian cancer: recent molecular insights and their impact on screening strategies. Curr Opin Oncol. 2011;23:526-30.
- Falconer H, Yin L, Grönberg H, et al. Ovarian cancer risk after salpingectomy: a nationwide population-based study. J Natl Cancer Inst. 2015;107(2).
- Havrilesky LJ, Gierisch JM, Moorman PG, et al. Oral contraceptive use for the primary prevention of ovarian cancer. Evid Rep Technol Assess (Full Rep). 2013;1-514.
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-15.
- Rizzuto I, Behrens RF, Smith LA. Risk of ovarian cancer in women treated with ovarian stimulating drugs for infertility. Cochrane Database Syst Rev. 2013;(8):CD008215.
- Baandrup L, Friis S, Dehlendorff C, et al. Prescription use of paracetamol and risk for ovarian cancer in Denmark. J Natl Cancer Inst. 2014;106(6):dju111.
Summary
Ovarian cancer develops due to a complex interplay between hereditary and environmental factors. Although often described as one disease, ovarian cancer is actually a group of distinct tumor types. Recent research has indicated that a large percentage of ovarian cancers may originate from the fallopian tube epithelium. Although most cancers develop in patients without a known hereditary syndrome, it is clear that the number of familial cancers is larger than previously supposed. The two most common hereditary syndromes where ovarian cancer can develop are hereditary breast ovarian cancer syndrome (HBOC) and Lynch syndrome.