![Figur 3. Mjältvävnad hos en 3-åring med sicklecellanemi som splenektomerats. Notera fibros och blodstockning i den röda pulpan (hematoxylin–eosin-färgning) [20].](https://lakartidningen.se/wp-content/uploads/EditorialFiles/AI/%5bD4AI%5d/2016-024_3_webb.jpg)
Akut mjältsekvestrering är en akut livshotande komplikation till sicklecellanemi. Tillståndet är viktigt att känna till eftersom det kan vara den första kliniska manifestationen av sicklecellanemi.
Akut mjältsekvestrering är en av de vanligaste dödsorsakerna hos barn med sicklecellanemi och beror på att blod plötsligt ansamlas i mjälten med splenomegali, akut anemi och hypovolemisk chock som följd. Akut transfusion med erytrocytkoncentrat är ofta livräddande.
Det finns stark evidens för att alla barn med sicklecellanemi ska behandlas med hydroxiurea och skötas av multidisciplinära team. Allogen hematopoetisk stamcellstransplantation är den enda kurativa behandlingen, och indikationen har vidgats de senaste åren.
Incidensen i Sverige kommer troligtvis stiga med ökad migration. Här ges en översikt av tillståndet med svenska rekommendationer.
Sicklecellanemi rapporterades första gången 1911 av amerikanen Herrick, som beskrev abnorma blodkroppar hos en tandläkarstudent från Västindien med recidiverande smärta, ikterus och bensår [1]. År 1949 publicerades den berömda studien »Sickle cell anemia, a molecular disease« av Linus Pauling et al [2], där det för första gången kunde visas att ett avvikande protein var sjukdomsalstrande. Det öppnade därmed fältet för framtidens molekylärbiologi.
Några år senare, 1956, beskrev Ingram att tillståndet orsakas av en mutation i betaglobingenen på kromosom 11, vilket resulterar i att den 6:e aminosyran är utbytt från glutamin till valin [3].
I denna artikel ges en översikt av sicklecellanemi och den akuta livshotande komplikationen mjältsekvestrering hos barn (Fakta 1), exemplifierat med en fallbeskrivning (Fakta 2).
Epidemiologi
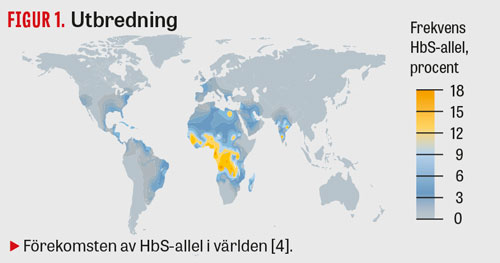
Sicklecellanemi är den vanligaste hemoglobinopatin i världen; uppskattningsvis föds ca 5 476 000 (5 291 000–5 679 000) heterozygota barn och 312 000 (294 000–330 000) homozygota barn i världen varje år [4]. 80 procent av dessa barn föds i låg- till medelinkomstländer [5]. Sjukdomen förekommer framför allt i områden där malaria är eller har varit endemisk (Figur 1), eftersom heterozygota bärare anses ha en överlevnadsfördel vid malaria [6].
Utan adekvat behandling, vilken sällan finns tillgänglig i utvecklingsländer, dör många av barnen före 5 års ålder [6]. Tidig diagnostik minskar risken för allvarliga komplikationer och död [7], och i många utvecklade länder finns numera nyföddhetsscreening.
I Sverige är prevalensen okänd, och det finns ingen screening för sicklecellhemoglobin/sicklecellanemi. Eftersom antalet människor i Sverige med härkomst från områden med sicklecellanemi ökar, finns det skäl att överväga om någon form av riktad screening bör införas.
Patofysiologi
Sicklecellanemi är en autosomalt recessiv hemoglobinopati som beror på syntes av defekt hemoglobin. Under fosterlivet består hemoglobinet (HbF) av två α- och två γ-globinkedjor (α2γ2). Efter födseln sker en gradvis övergång till produktion av adult hemoglobin (HbA) som består av två α- och två β-kedjor (α2β2). Mutationen på kromosom 11 förorsakar en förändrad β-globinkedja. βS bildas i stället för den normala β-kedjan, vilket ger upphov till sicklecellhemoglobin, HbS (α2βS2).
Den vanligaste varianten av sjukdomen uppträder hos individer som är homozygota för sicklecellgenen; de uppvisar vanligtvis ca 90 procent HbS i blodet. Heterozygota bärare har ca 40 procent HbS, normal mängd hemoglobin i blodet och normala röda blodkroppskonstanter. Bärarskapet upptäcks således inte med vanlig blodprovstagning utan endast med analys av fraktionerat hemoglobin eller genotypning.
Heterozygota bärare, som i grunden bör betraktas som friska individer, har associerats med ökad risk för njursjukdom [8, 9] och kan under extrema yttre förhållanden (t ex lågt FiO2, höga atmosfärtryck) och mycket kraftig fysisk ansträngning också få episoder av sickling [10]. Om en heterozygot bärare samtidigt har en annan mutation i den andra hemoglobingenen, såsom HbC och β-talassemi [Sβ+ eller Sβ°]), får de emellertid hematologisk påverkan och ofta kliniska symtom på sicklecellsjukdom.
De allvarligaste formerna av sjukdomen är HbSS och HbSβ°, vilka kan vara svåra att skilja åt kliniskt. HbSC och HbSβ+ är som regel mindre allvarliga [11]. Sjukdomens svårighetsgrad bestäms även delvis av individens nedärvda förmåga att postnatalt bilda HbF, där högre halter av HbF är associerade med lindrigare symtom [12].
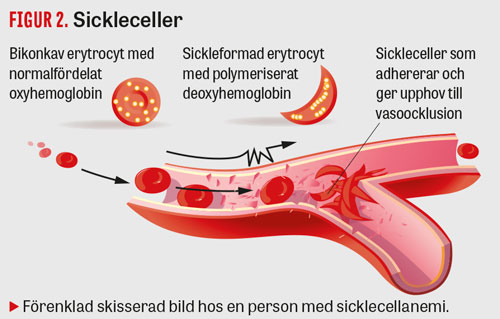
Risken för att HbS polymeriserar i erytrocyten ökar vid hypoxi, acidos och intracellulär dehydrering. Det resulterar i en formförändring av erytrocyten, vilken antar ett karakteristiskt långsträckt, månskäreformat utseende (Figur 2). Dessa s k sickleceller aggregerar med varandra, interagerar med endotelet [13] och ger upphov till ocklusion i mindre kärl (vasoocklusiv episod) [14].
Den uppkomna ischemin följd av reperfusion leder till en komplex kaskad där kombinationen hypoxi, inflammation, endotelskada, aktiverat koagulationssystem och minskad kväveoxidhalt leder till kronisk inflammation och vaskulopati [14-17]. Organ med långsamt blodflöde och låga syrgastryck som mjälte och benmärg är särskilt utsatta, liksom organ med begränsat blodflöde såsom näthinnan och delar av njuren samt proximala femur och humerus [18]. Processen med polymerisering är delvis reversibel, men vid upprepade episoder skadar den cellens cytoskelett, vilket ger upphov till irreversibel sickling [19]. Det ger förkortad livslängd hos erytrocyten med åtföljande kronisk hemolytisk anemi [11].
Pediatrisk symtomvariation
Sicklecellanemi manifesterar sig med olika symtom i olika åldrar. Det nyfödda barnet skyddas av den högre halten HbF i blodet, vilken varar ca 3 månader [11]. Snabb försämring av mjältfunktionen i kombination med att yngre barn generellt har låga nivåer av opsoniserande antikroppar ger kraftigt ökad infektionskänslighet [18]. Risken för invasiv pneumokocksjukdom är förhöjd 300–500 gånger [11]. Därför bör barn med sicklecellanemi få profylaktisk antibiotikabehandling omedelbart vid diagnos (oftast upp till 5 års ålder) och utökade vaccinationer med konjugerat pneumokockvaccin från 2 månaders ålder, 23-valent vaccin vid 2 års ålder (booster vid 5 år) och influensavaccin årligen [18, 20].
Den reducerade livslängden av röda blodkroppar med HbS (10–20 dygn) leder till anemi, som kan noteras redan vid 3–6 månaders ålder och leder till kompensatorisk benmärgshyperplasi. Smärtsamma inflammationer i fingrar och tår (daktylit), utlösta av vasoocklusion i metakarpal- och metatarsalbenen, är också vanliga i denna ålder.
Från ca 1 års ålder tillkommer risk för smärtattacker från rörbenen, obstruktion av övre luftvägarna samt stroke och från ca 4 års ålder även akut bröstsyndrom, sekvestrering av blod i levern och priapism [17].
Mjälten och sicklecellanemi
Mjälten är det första organ som tar skada av sicklecellanemi, och hyposplenism är vanligt redan vid 3–12 månaders ålder [20-22]. Mjälten tar emot ca 3–5 procent av kroppens blodvolym [19], och dess unika mikroanatomi och långsamma mikrocirkulation gör att blodburna partiklar, antigener och skadade erytrocyter dynamiskt filtrerats innan de fagocyteras.
De histopatologiska förändringarna vid sicklecellanemi i mjälten beskrevs redan 1935, men patofysiologin är fortfarande inte fullständigt klarlagd [23]. Man misstänker att sicklecellerna fastnar i mjältens matrix på grund av ökat antal adhesionsmolekyler och aktivering av specifika proteiner [20].
Hos äldre barn med sicklecellanemi sker en intermittent intrasplenisk sickling med lokala små infarkter med ärrbildning och fibros som följd (Figur 3), vilket till slut leder till en icke-funktionell mjälte (funktionell aspleni eller autosplenektomi) [20].
Vid 5 års ålder uppskattas ca 90 procent ha funktionell aspleni, vilket sannolikt är anledningen till att mjältsekvestrering sällan sker efter denna ålder [24].
Mjältsekvestrering
Akut mjältsekvestrering är den tidigaste livshotande komplikationen hos patienter med sicklecellanemi; den yngsta patient som beskrivits var endast 5 veckor gammal [25]. Efter infektion är det den vanligaste dödsorsaken hos barn under 10 års ålder med sicklecellanemi [26]. Bland de kliniskt lindrigare genotyperna HbSC och HbSβ+ är förlusten av mjältfunktion långsammare, och sekvestrering kan ske så sent som i tonåren eller i vuxen ålder. Akut mjältsekvestrering hos äldre barn är ofta smärtsam på grund av samtidig mjältinfarkt [11].
Utlösande faktorer är dåligt kända, även om infektion och feber tros kunna öka erytrocyternas sickling i den röda pulpan.
Erytrocyterna ansamlas (sekvestrera = beslagta) i mjälten med akut ökande splenomegali och sjunkande hemoglobinhalt följt av cirkulationspåverkan med hypovolemisk chock. Splenomegalin kan vara mycket uttalad, och det är den hastiga ökningen av mjältstorleken som är typisk för tillståndet eftersom en del barn med sicklecellanemi har kronisk splenomegali.
Akut mjältsekvestrering kan leda till döden inom några timmar och kräver snabb behandling [20, 26]. Definitionsmässigt föreligger tillståndet vid splenomegali och minskning av hemoglobinvärdet med minst 20 g/l från ursprungsvärdet samt tecken till benmärgsaktivitet med normala eller förhöjda retikulocyter [26]. Det sistnämnda är för att kunna differentiera mot aplastisk kris utlöst av parvovirus B19, som vid sicklecellanemi också kan ge akut benmärgshämning med snabbt sjunkande hemoglobin.
I två retrospektiva kohortstudier från USA och Brasilien följdes barn med HbSS eller HbSβ°. De flesta diagnostiserades i samband med nyföddhetsscreening. Uppföljningstiden var 2 till 10 år. Under denna tid fick 29–31 procent av barnen minst en episod av akut mjältsekvestrering, merparten före 2 års ålder. Risken för recidiv var 49–67 procent [27]. 11 av de totalt 13 fallen med dödlig utgång inträffade vid den första episoden av mjältsekvestrering och i samtliga fall innan blodtransfusion hann ges.
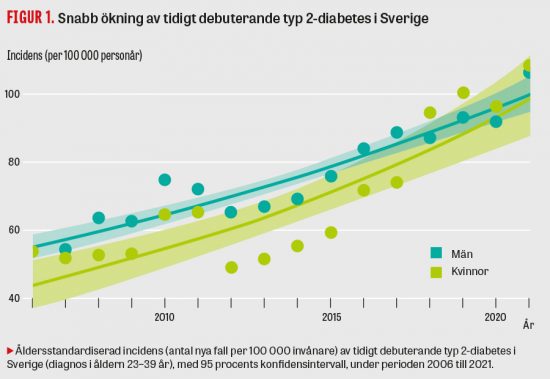
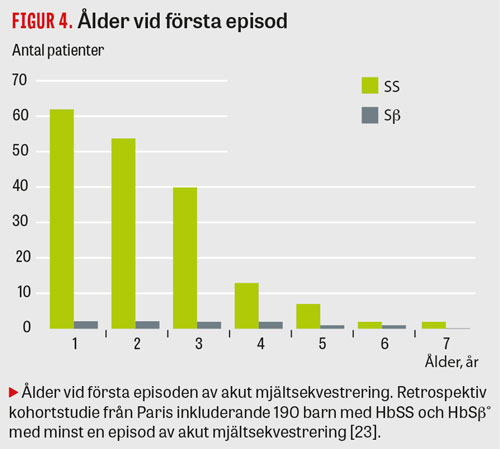
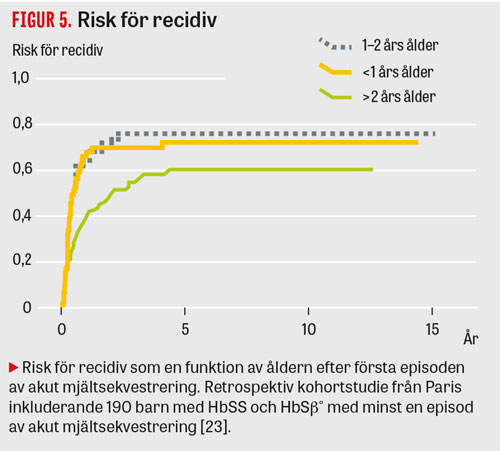
I en kohortstudie från Frankrike med 190 barn med känd HbSS och HBSβ hade 12,6 procent akut mjältsekvestrering med en recidivfrekvens av 67 procent. Åldern var den enda faktorn som påverkade risken för återfall (Figur 4 och 5) [23].
Akut behandling
Tillståndet bör övervägas och behandlas vid splenomegali, blekhet och hemodynamisk instabilitet i kombination med anemi. Behandlingen består av att omedelbart korrigera hypovolemin med kristalloider 10–20 ml/kg. Merparten av alla episoder av akut mjältsekvestrering kräver snabb blodtransfusion. Om Hb-värdet är <60 g/l ges erytrocytransfusion motsvarande 10 ml/kg, med målet att transfundera till patientens basnivå eller om denna är okänd inte höja Hb över 80 g/l.
Över denna nivå ökar risken för komplikationer som inträffar när tillståndet reverseras och de i mjälten ansamlade erytrocyterna snabbt återvänder till blodbanan, s k autotransfusion. Detta kan leda till hyperviskositet och risk för stroke [18]. 10 ml/kg erytrocytkoncentrat höjer Hb ca 25 g/l och erytrocytvolymfraktionen (EVF) med ca 7–9 enheter [28]. Man bör också kontrollera trombocytkoncentrationen (TPK), eftersom TPK-värdet kan sjunka avsevärt till följd av splenomegalin [18].
Radiologisk undersökning bör inte fördröja behandlingen. Tillståndet kan vid den initiala handläggningen vara svårt att skilja från sepsis och akut blödning, och eftersom infektion också kan vara utlösande orsak till akut mjältsekvestrering ska antibiotika med aktivitet mot kapslade bakterier ges intravenöst. Kraftig retikulocytos i kombination med undersökning av erytrocytmorfologi är till stor hjälp i differentialdiagnostiken.
I enstaka mycket svåra fall kan akut splenektomi vara nödvändig.
Sekundärprofylax och uppföljning
Eftersom tillståndet är potentiellt livshotande och risken för recidiv är hög, är det viktigt att försöka förhindra nya episoder. Alla föräldrar till barn med sicklecellanemi bör vid diagnostillfället få information om akut mjältsekvestrering där de uppmanas att söka akut vård vid snabbt påkommen blekhet och försämrat allmäntillstånd [20]. Föräldrarna bör även undervisas i att palpera mjältstorleken. Mortaliteten har sjunkit markant [29] sedan neonatal screening för sicklecellanemi införts i bl a Storbritannien, Holland och delar av USA och dessa barn inkluderats i vårdprogram och föräldrarna undervisats avseende symtom på mjältsekvestrering [23].
Förutom allogen stamcellstransplantation finns två etablerade metoder att minska eller eliminera risken för ny episod av akut mjältsekvestrering: kronisk transfusionsbehandling och splenektomi.
Kronisk transfusionsbehandling minskar risken för recidiv och används framför allt hos barn med hög strokerisk [30]. Upprepade blodtransfusioner har flera nackdelar. Alloimmunisering kan uppstå även om risken i dag är lägre när utvidgad fenotypning av blod görs. En del patienter som avslutar behandlingen med regelbundna transfusioner utvecklar kort därefter akut mjältsekvestrering [31]. Risken för biverkningar (framför allt järninlagring) gör att man sällan väljer tranfusionsbehandling.
Splenektomi eliminerar risken för akut mjältsekvestrering, men eftersom barnen som regel är unga i samband med sin första episod väljer många att behandla med splenektomi först efter den andra episoden på grund av oro för ökad risk för sepsis med kapslade bakterier efter splenektomi. Eftersom barn med sicklecellanemi redan från ung ålder har kraftigt nedsatt mjältfunktion, är det dock osäkert om risken för sepsis ytterligare höjs. Flera studier visar också att risken för infektion inte ökar efter splenektomi om utökad vaccination och antibiotikaprofylax ges [32, 33].
I en studie från Jamaica av 130 patienter som splenektomerats under en period av 22,5 år noterades ingen statistiskt signifikant skillnad avseende mortalitet eller episoder med sepsis och bakteriemi jämfört med kontrollgruppen. Däremot noterades ett ökat antal smärtsamma kriser och akut bröstsyndrom. Det kan indikera att mjältkomplikationer kan vara en prediktor för generell sjukdomssvårighet [32].
Andra fördelar med splenektomi är att barn som tidigare behandlats med blodtransfusioner ofta får minskat transfusionsbehov och sjunkande halt av HbS, sannolikt på grund av förlängd överlevnad av de transfunderade erytrocyterna [34].
Sammantaget varierar behandlingsmetoderna i världen efter första episoden av akut mjältsekvestrering, sannolikt på basis av bristande evidens för olika behandlingsregimer. I svenska riktlinjer rekommenderas att splenektomi övervägs redan efter första episoden [18].
För att minska risken för akuta och sena komplikationer bör barn med sicklecellanemi regelbundet kontrolleras hos ett pediatriskt behandlingsteam med erfarenhet av sjukdomen. Det finns i dag starka belägg för att alla barn med sicklecellanemi bör behandlas med hydroxiurea från tidig ålder [35, 36]. Dock finns inga studier som visar att denna behandling minskar risken för akut mjältsekvestrering, men den kan eventuellt ha en viss positiv inverkan på mjältfunktionen [35].
Allogen hematopoetisk stamcellstransplantation är i dagsläget den enda kurativa behandlingen. I takt med att resultaten för denna behandling förbättrats har indikationerna, som baseras på en bedömning av den individuella risken för allvarliga komplikationer av sicklecellanemi, utvidgats de senaste åren. Alla barn med sicklecellanemi bör utvärderas med avseende på indikation för allogen hematopoetisk stamcellstransplantation. Vid vissa centra erbjuds behandlingen i dag till alla barn med sicklecellanemi som har ett HLA-identiskt syskon. För barn med ökad risk för allvarliga sicklecellrelaterade komplikationer bör indikation med alternativ donator som orelaterad HLA-identisk donator eller navelsträngsblod övervägas [37, 38].
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 2. Fallbeskrivning
Fallet rör en tidigare frisk svenskfödd 4-årig pojke. Föräldrarna härstammade från Mellanöstern och han var fjärde barnet av fem syskon. Pojken hade haft ett dygns feber med relativt gott allmäntillstånd, men blev under andra dagen alltmer allmänpåverkad.
Vid ankomst till akutmottagningen bars han in av sina föräldrar, kraftigt blek och allmänpåverkad. I status noterades förhöjd kroppstemperatur (38,1 °C), sänkt syremättnad (81 procent), uttalad blekhet, takykardi och förlängd kapillär återfyllnad.
Pojken hade uttalad mjältförstoring (9 cm nedom arcus), måttlig lymfadenopati på halsen och var misstänkt nackstel. Inga petekier eller andra utslag noterades.
Han chockbehandlades med intravenös vätska samt fick cefotaxim intravenöst efter blododling.
Akuta laboratorieanalyser visade Hb 26 g/l, anisocytos, TPK 53 × 109/l och LPK 16 × 109/l med morfologiskt avvikande lymfocyter. CRP var 19 mg/l, ASAT lätt förhöjt (5,86 µkat/l), men ALAT normalt.
Pojken transfunderades med 8 ml/kg 0 Rh-negativt blod och överfördes till barnintensivvårdsavdelning. Där vårdades han kortvarigt innan han kunde flyttas till barnonkologisk enhet för vidare utredning av misstänkt leukemi.
Hb-värdet stabiliserades kring 90 g/l, och TPK-värdet steg långsamt. Pojken hade uttalad retikulocytos, 422 × 109/l, samt lätt förhöjt bilirubin, 39 µmol/l.
Lungröntgen visade förstorat hjärta och en liten mängd pleuravätska. Ultraljudsundersökning av buken visade uttalad mjältförstoring och möjligen viss leverförstoring. Hepatit-, hiv- och HTLV (humant T-lymfotropt virus)-serologi var negativ. Benmärgsundersökning efter två dygn uteslöt akut leukemi.
Vid utvidgad anamnes framkom ingen konsangvinitet, och föräldrar och syskon var friska utan känd anemi. Det framkom att pojken vid minst två tillfällen sökt vård akut på grund av smärta från leder utan känt trauma.
Blodtransfusionen vid det akuta insjuknandet försvårade diagnostiken, dock kunde man, på blod som sparats på laboratoriet före transfusionen, analysera fraktionerade hemoglobiner. Analysen visade 66 procent HbS förenligt med antingen homozygoti för HbS eller kombinerad heterozygoti för HbS och β°-talassemi.
Genotypning påvisade två HbS-alleler och bekräftade homozygoti för HbS (dvs sicklecellanemi). Pojken fick behandling med antibiotikaprofylax, hydroxiurea och folacin.
6 månader efter episoden insjuknade han i en vasoocklusiv episod med feber, buksmärtor, ledvärk och anemi med Hb 44 g/l. I efterförloppet utreddes han på grund av svår sjukdom för indikation för stamcellstransplantation. Pojkens syskon var HLA-identiskt, och han har nu genomgått allogen stamcellstransplantation.
Fallet illustrerar det fulminanta och allvarliga förloppet av akut mjältskvestrering. Den svåra anemin i kombination med samtidig retikulocytos talade emot leukemi; sicklecellanemi eller någon annan form av hemolytisk anemi var mer trolig.
Familjen hade inget känt påbrå från Afrika, vilket illustrerar vikten av att känna till att tillståndet förekommer utanför detta område (Figur 1). Det är troligt att incidensen i Sverige kommer att stiga med ökad migration.
Referenser
- Savitt TL, Goldberg MF. Herrick’s 1910 case report of sickle cell anemia. The rest of the story. JAMA. 1989;261:266-71.
- Pauling L, Itano HA, et al. Sickle cell anemia, a molecular disease. Science. 1949;110:543-8.
- Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326-8.
- Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381:142-51.
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331-6.
- Williams TN, Obaro SK. Sickle cell disease and malaria morbidity: a tale with two tails. Trends Parasitol. 2011;27:315-20.
- Panepinto JA, Magid D, Rewers MJ, et al. Universal versus targeted screening of infants for sickle cell disease: a cost-effectiveness analysis. J. Pediatr. 2000;136:201-8.
- Ataga KI, Orringer EP. Renal abnormalities in sickle cell disease. Am J Hematol. 2000;63:205-11.
- Naik RP, Derebail VK, Grams ME, et al. Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. JAMA. 2014;312:2115-25.
- Key NS, Connes P, Derebail VK. Negative health implications of sickle cell trait in high income countries: from the football field to the laboratory. Br J Haematol. 2015;170:5-14.
- Quinn CT. Sickle cell disease in childhood: from newborn screening through transition to adult medical care. Pediatr Clin North Am. 2013;60:1363-81.
- Powars DR, Weiss JN, Chan LS, et al. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921-6.
- Hebbel RP, Boogaerts MA, Eaton JW, et al. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med. 1980;302:992-5.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762-9.
- Sugihara K, Sugihara T, Mohandas N, et al. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood. 1992;80:2634-42.
- Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343-60.
- Steinberg MH. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. ScientificWorldJournal. 2008;8:1295-324.
- Svenska barnläkarföreningen, Vårdplaneringsgruppen för pediatrisk hematologi. Vårdprogram Sicklecellanemi. 2006. http://www.blf.net/onko/page4/page29/files/SCA%20VPH%202006%20uppdat%202012.pdf
- Lux SE, John KM, Karnovsky MJ. Irreversible deformation of the spectrin-actin lattice in irreversibly sickled cells. J Clin Invest. 1976;58:955-63.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol. 2014;166:165-76.
- Rogers ZR, Wang WC, Luo Z, et al. Biomarkers of splenic function in infants with sickle cell anemia: baseline data from the BABY HUG Trial. Blood. 2011;117:2614-7.
- William BM, Corazza GR. Hyposplenism: a comprehensive review. Part I: basic concepts and causes. Hematology. 2007;12:1-13.
- Brousse V, Elie C, Benkerrou M, et al. Acute splenic sequestration crisis in sickle cell disease: cohort study of 190 paediatric patients. Br J Haematol. 2012;156:643-8.
- Brown AK, Sleeper LA, Miller ST, et al. Reference values and hematologic changes from birth to 5 years in patients with sickle cell disease. Cooperative Study of Sickle Cell Disease. Arch Pediatr Adolesc Med. 1994;148:796-804.
- Airede AI. Acute splenic sequestration in a five-week-old infant with sickle cell disease. J Pediatr. 1992;120:160.
- Topley JM, Rogers DW, Stevens MC, et al. Acute splenic sequestration and hypersplenism in the first five years in homozygous sickle cell disease. Arch Dis Child. 1981;56:765-9.
- Emond AM, Collis R, Darvill D, et al. Acute splenic sequestration in homozygous sickle cell disease: natural history and management. J Pediatr. 1985;107:201-6.
- Svenska barnläkarföreningen, Vårdplaneringsgruppen för pediatrisk hematologi. Transfusion av blodkomponenter till barn. Aktuella rekommendationer från VPH. 2011. http://www.blf.net/onko/page4/page29/files/Transfusions_pm_Barn_2011.pdf
- Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92:905-12.
- Rao S, Gooden S. Splenic sequestration in sickle cell disease: role of transfusion therapy. Am J Pediatr Hematol Oncol. 1985;7:298-301.
- Kinney TR, Ware RE, Schultz WH, et al. Long-term management of splenic sequestration in children with sickle cell disease. J Pediatr. 1990;117:194-9.
- Wright JG, Hambleton IR, Thomas PW, et al. Postsplenectomy course in homozygous sickle cell disease. J Pediatr. 1999;134:304-9.
- Vick LR, Gosche JR, Islam S. Partial splenectomy prevents splenic sequestration crises in sickle cell disease. J Pediatr Surg. 2009;44:2088-91.
- Haricharan RN, Roberts JM, Morgan TL, et al. Splenectomy reduces packed red cell transfusion requirement in children with sickle cell disease. J Pediatr Surg. 2008;43:1052-6.
- Wang WC, Ware RE, Miller ST, et al. A multicenter randomised controlled trial of hydroxyurea (hydroxycarbamide) in very young children with sickle cell anaemia. Lancet. 2011;377:1663-72.
- McGann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf. 2015;14:1749-58.
- Walters MC. Update of hematopoietic cell transplantation for sickle cell disease. Curr Opin Hematol. 2015;22:227-33.
- Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99:811-20.
Summary
Acute splenic sequestration in children with sickle cell disease – an overview
Acute splenic sequestration (ASS) is a life-threatening complication of sickle cell disease (SCD). The condition is important to recognize due to the fact that it can occur with previously unknown disease. ASS is one of the most common causes of death in children with SCD and is the result of blood suddenly getting congested in the spleen, resulting in splenomegaly, acute anemia, and hypovolemic shock. Timely and appropriate treatment is essential in preventing death. Episodes of ASS before one year of age are associated with a higher risk of recurrence. There is no established effective treatment for recurrent ASS; however, there is evidence that all children with SCD should be treated with hydroxyurea. In Sweden, our recommendation is to evaluate the indications for splenectomy after the first episode of ASS. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative treatment, and all children with SCD should be evaluated with regard to the potential success of HSCT. This article presents an overview of the condition with Swedish recommendations.