Sammanfattat
Hereditär hemokromatos är en vanlig ärftlig sjukdom med en prevalens i Sverige på cirka 0,5 procent.
Sjukdomen är oftast asymtomatisk, men de vanligaste symtomen är trötthet, ledbesvär (i främst metakarpofalangeal- och fotleder) och leverpåverkan. Cirka 5 procent av obehandlade patienter utvecklar levercirros.
Sjukdomen orsakas av mutationer i HFE-genen, vilket leder till brist på det järnreglerande hormonet hepcidin, med ett ökat järnupptag från tunntarmen som följd.
Misstänk hemokromatos vid leverpåverkan utan annan förklaring, oklara ledbesvär eller förhöjd järnmättnad i plasma.
Diagnosen ställs med HFE-mutationsanalys, som visar homozygoti för mutationen C282Y eller sammansatt heterozygoti för C282Y/H63D.
Leverbiopsi behöver endast göras vid ferritin >1 000 µg/l, då det finns risk för leverskador, eller i de fall då HFE-mutation saknas.
Behandlingen är blodtappningar (venesectio) på 400–500 ml blod per vecka tills ferritinvärdet normaliseras.
Underhållsbehandlingen, som är livslång, innebär regelbundna blodtappningar 2–6 gånger per år, där tappningsfrekvensen ska styras av ferritinvärdet, som bör ligga kring 50–100 µg/l.
Hereditär hemokromatos är en vanlig ärftlig sjukdom i Sverige som kan leda till levercirros om den inte upptäcks eller behandlas i tid. Den upptäcks oftast på grund av förhöjda levervärden eller förhöjd järnmättnad och förhöjt ferritin [1]. Sjukdomen leder till allvarlig organskada hos 5 procent av personer med den ärftliga formen, och eftersom det finns effektiv behandling bör sjukdomen upptäckas och behandlas i tid.
Förhöjt serumferritin är ett vanligt problem i den kliniska vardagen. Andra orsaker till en ferritinstegring förutom hereditär hemokromatos är inflammation, leverpåverkan, alkoholöverkonsumtion eller dysmetabolt järnöverskott [2]. I dessa fall beror det stegrade serumferritinet på läckage från påverkade leverceller, underliggande inflammation, där ferritin verkar som en akutfasreaktant, eller metabola syndromet, där orsaken till ferritinstegringen är okänd.
Etiologi och patogenes
De senaste 15 åren har givit helt ny och omvälvande kunskap om järnomsättningen [3-5]. Hereditär hemokromatos beror i de allra flesta fall på en mutation i HFE-genen (C282Y). HFEgenens protein reglerar syntesen av ett hormon, hepcidin, som i sin tur styr kroppens järnupptag. HFE-proteinet är lokaliserat på levercellernas cellyta, där det interagerar med transferrinreceptor 1 (TfR1) och 2 (TfR2). TfR1 binder järn-transferrin, vilket minskar affiniteten mellan HFE och TfR1. När serumjärn är högt släpper alltså HFE-proteinet från TfR1 och binder i stället TfR2 i ett komplex. När detta komplex aktiveras skickas signaler till cellkärnan att öka syntesen av järnhormonet hepcidin. Både mutationer i HFE, som är vanliga i norra Europa, och i TfR2, som beskrivits från medelhavsområdet, leder därför till sänkt hepcidinsyntes.
Hepcidin utsöndras från levercellen och sänker serumjärn genom att blockera järnupptaget från tarmcellerna och frisättningen av järn från makrofagerna. Hepcidinbristen vid hemokromatos leder till fortsatt högt järnupptag. Järnupptaget från tarmen stimuleras, järnfrisättningen till plasma ökar och järnmättnaden i plasma stiger. Högt serumjärn eller hög järnmättnad är det viktigaste kliniska tecknet vid hemokromatos.
Järnpoolen i plasma är liten, endast 20 mg järn. Totalt finns ca 4,5 g järn i kroppen, varav 0,5 g utgörs av en reserv. Hos patienter som är homozygota för C282Y-mutationen i HFE-genen och har förhöjt ferritin brukar det finnas från 2 g upp till ibland 50 g järn inlagrat i kroppens parenkymatösa organ.
Järnpoolen har hög omsättning, där merparten transporteras till benmärgen för produktion av röda blodkroppar. Döda erytrocyter fagocyteras av makrofager, som frisätter järnet till plasmapoolen. Hepcidin hämmar flödet av järn in i denna järnpool från både tarmceller och makrofager. Vid inflammation stimuleras hepcidinsyntesen av interleukin-6. Den höga hepcidinaktiviteten vid inflammation leder till att frisättningen av järn till järnpoolen stryps, och serumjärn sjunker snabbt, redan inom några timmar. Järnmetabolismen vid inflammation och hemokromatos är alltså två motpoler som beror på ökad respektive minskad hepcidinaktivitet i plasma.
Incidens och penetrans
Den vanligaste orsaken till genetisk hemokromatos är mutationen C282Y i HFE-genen. Denna mutation förekommer hos ca 1/15 av den svenska befolkningen i heterozygot form och hos ca 1/200 i homozygot form [5]. Den är betydligt vanligare än andra ärftliga leversjukdomar. Cirka 5 procent får en allvarlig fenotyp med stora mängder inlagrat järn. Det är således 1/4 000 svenskar som får en allvarlig form av sjukdomen. Det är vanligare med symtom hos män än hos kvinnor. Man anser att mutationen i HFE-genen, som ligger i HLA-regionen, som är ett mycket stabilt område, uppstod hos en keltisk anfader för ca 1 500 år sedan. Den är vanlig i nordvästra Frankrike, Storbritannien och Irland, men spreds under vikingatiden till Skandinavien. Sjukdomen är ovanligare i södra Europa och sällsynt hos asiater och afrikaner.
En annan mutation, H63D, kan i kombination med C282Y leda till lindrig hemokromatos. Denna genotyp kallas »compound« (sammansatt) heterozygoti. Homozygoti för enbart H63D leder inte till hemokromatos. Heterozygoti för H63D är mycket vanligt och ses hos 15 procent av jordens befolkning. Av svenska patienter med hereditär hemokromatos har ca 90 procent homozygoti för C282Y och 5 procent sammansatt heterozygoti för C282Y och H63D [5]. Således saknar 5 procent av hemokromatospatienterna dessa mutationer. Mutationer i andra järnreglerande gener har beskrivits vid hemokromatos, t ex TfR2 och hemojuvelin. Hemojuvelin är en annan regulator av hepcidinsyntesen och har fått sitt namn av att mutationer i denna gen kan leda till juvenil hemokromatos med debut i 20-årsåldern. Juvenil hemokromatos är extremt sällsynt, och inget fall har beskrivits i Sverige.
Även om hemokromatos kan leda till leverpåverkan och levercirros upptäcks de flesta hemokromatospatienterna vid rutinmässig provtagning på grund av förhöjda järnparametrar i serum vid en hälsokontroll eller vid utredning av förhöjda aminotransferaser. Bärarskap av genotyperna C282Y/C282Y eller C282Y/H63D i HFE-genen ses hos 0,5 procent av den svenska befolkningen. Cirka 70 procent av C282Y-homozygoter utvecklar förhöjd järnmättnad i serum med eller utan ferritinstegring, så den kliniska penetransen är inte hundraprocentig.
Vid ökande järninlagring stiger serumferritin. Aminotransferaserna börjar stiga vid ett serumferritin över 500–900 µg/l. Patienter med ferritinvärden >1 000 µg/l kan utveckla levercirros, diabetes, hypogonadism, hjärtarytmier eller hypofysinsufficiens. Levercirros sågs tidigare hos 20 procent av de diagnostiserade patienterna. Vid etablerad cirros är risken att utveckla hepatocellulär cancer 2–3 procent per år.
Diagnostik
De flesta patienter med hemokromatos är symtomfria, och när symtom uppträder är de oftast ospecifika. Det vanligaste symtomet som patienterna beskriver är en generell trötthet. Speciellt män med hemokromatos kan utveckla artropati med stelhet och uppdrivningar i småleder, främst metakarpofalangeal- (MCP) eller fotleder (Figur 1), vilket ses hos upp till av 30 procent av alla som diagnostiseras. Ledbesvär i MCP- eller fotleder hos män ska därför utredas med avseende på hemokromatos.
Vanligen hittar man patienterna på grund av höga levervärden eller ett högt serumjärn, hög järnmättnad och högt serumferritin. Vid förhöjd järnmättnad (över 55 procent) i två upprepade prov finns en klar indikation för HFE-mutationsanalys oavsett ferritinvärde eftersom det är ett kardinaltecken på hereditär hemokromatos. Om HFE-testet visar homozygoti för C282Y eller sammansatt heterozygoti för C282Y/H63D är diagnosen klar [6].
Om HFE-testet däremot visar normalfynd krävs leverbiopsi för att påvisa ökad mängd järn i levern [7]. Järnfärgning av histologiska snitt påvisar ett blått järnpigment i de leverceller som ligger runt leverns portazoner. Vid avancerad hemokromatos ses järn även i Kupffer- och gallgångsceller (Figur 2). Olika grader av fibros ses ofta. Inflammation brukar saknas helt eller så är den lindrig. Vid sekundär hemokromatos ses järn främst i makrofager (Kupfferceller). Vid dysmetabolt järnöverskott (t ex metabolt syndrom, fettlever eller typ 2-diabetes) ses ibland en lindrig järninlagring i hepatocyter och samtidig steatos (Figur 3). Vid järnöverskott orsakat av alkohol eller kronisk hepatit C ses en måttlig järninlagring i hepatocyter och ofta en något mer uttalad järninlagring i Kupfferceller på grund av samtidig cellnekros med fagocytos av järninnehållande cellrester.
Vid kontraindikaton för leverbiopsi kan järnmängden i levern uppskattas semikvantitativt med MR-undersökning [8] och fibrosstadium mätas med elastografi [9]. Vid MR-undersökningen ses förhöjd järnhalt i levern, men inte i mjälten, eftersom retikuloendoteliala celler är järnfattiga vid hereditär hemokromatos. Om serumferritin är över 1 000 µg/l rekommenderas leverbiopsi (eller elastografi) även för patienter som har HFE-mutationen eftersom det är vid denna nivå det föreligger risk för levercirros.
Differentialdiagnoser
En differentialdiagnos till hereditär hemokromatos är sekundärt järnöverskott, som ses hos talassemipatienter som har behandlats med upprepade blodtransfusioner eller hos patienter med renal anemi som behandlas med parenteralt järn. I dessa fall erbjuder differentialdiagnostiken sällan några problem. Svårare kan det vara att skilja patienter med dysmetabolt järnöverskott, porphyria cutanea tarda, alkoholleversjukdom, kronisk hepatit C eller levercirros av annan genes från dem med hereditär hemokromatos. Även dessa patienter har ofta förhöjt serumferritin och ibland leverpåverkan, och de kan också utveckla ett lindrigt eller måttligt järnöverskott i levern.
Också vid alkoholleversjukdom, cirros och hepatit C är patogenesen hepcidinbrist eftersom både alkohol och leverinflammation hämmar hepcidinsyntesen i levern. Orsaken till järninlagringen vid dysmetabolt järnöverskott är okänd. Eftersom genetisk hemokromatos är vanlig måste man komma ihåg att den kan vara en skadlig ko-faktor vid samtidig annan leversjukdom [11]. Om det föreligger ett högt serumferritin bör man således överväga om patienten kan ha både primär leversjukdom och genetisk hemokromatos.
Behandling
Man brukar alltid behandla patienter med ferritinstegring om de har mutationerna C282Y/C282Y eller C282Y/H63D i HFE-genen i syfte att förebygga sjukdomsutveckling [12, 13]. Behandlingen utgörs av blodtappningar (venesectio). Under den initiala intensivbehandlingen tappar man i normalfallet 450–500 ml varje vecka. Patienter med hereditär hemokromatos tolererar vanligtvis blodtappning mycket bra.
Vid eventuell intolerans mot frekvent tappning kan man i stället göra feres och ge tillbaka plasman. I en nyligen publicerad studie visar man att detta även kan vara bättre än ren venesektion [14]. B-hemoglobin kan sjunka något, speciellt de första 3–4 veckorna innan erytropoetinsyntesen ökat, men sällan under 120 g/l. Regelbunden blodtappning stimulerar erytropoesen, vilket mobiliserar överskottsjärnet från levern. Vid höga ferritinvärden kan intensivbehandlingen pågå i över ett år, ibland flera år, men det vanliga är 4–6 månader.
Man brukar avsluta intensivbehandlingen när serumferritin närmar sig 50 µg/l. Efter avslutad intensivbehandling rekommenderas underhållsbehandling för att hålla serumferritin kring 30–100 µg/l. I normalfallet underhållsbehandlar man med blodtappningar 3–4 gånger per år hos kvinnor och 4–6 gånger per år hos män. Under den livslånga underhållsbehandlingen räcker det att man kontrollerar serumferritin en gång per år, varefter det kommande årets tappningsintervall justeras beroende på om värdet stigit eller sjunkit.
Artropatin kan endast behandlas symtomatiskt med exempelvis antiflogistika eller proteskirurgi – om det blir nödvändigt [10]. Tyvärr lindras varken smärtan eller ledstelheten av blodtappningarna. Om levercirros har utvecklats bör patienten följas med ultraljudsundersökningar, vanligen två gånger per år, med tanke på den förhöjda risken för hepatocellulär cancer. Man bör även genomföra gastroskopi för att utesluta portal hypertension och utveckling av esofagusvaricer. Eventuell diabetes behandlas på sedvanligt sätt. Ibland kan insulinbehovet minska efter avslutad intensivbehandling med blodtappningar.
Prognos
Prognosen är mycket god om sjukdomen upptäcks i tid, innan organskador har hunnit utvecklas [12, 13, 15]. Då är överlevnaden i nivå med normalbefolkningens. Viss regress av bindvävsinlagringen i levern har beskrivits efter avslutad intensivbehandling hos patienter med fibros eller tidig cirros. Vid etablerad cirros ses ökad mortalitet till följd av cirroskomplikationerna och den förhöjda risken att utveckla hepatocellulär cancer.
Släktutredning
Eftersom sjukdomen ärvs autosomalt recessivt har syskon till en hemokromatospatient 25 procents risk att ha ärvt anlaget. Man brukar därför screena syskon till hemokromatospatienter med kontroll av järnmättnad och serumferritin. Om patienten är homozygot för C282Y-mutationen i HFE kan screening också göras med HFE-mutationsanalys. Barn till en förälder med homozygot sjukdom har statistiskt sett en risk på 1/30 att själva vara homozygota bärare av mutationen om båda föräldrarna har svenskt ursprung. Risken är således liten, men många föräldrar vill att barnen kontrolleras.
Sammanfattning
Ökad medvetenhet om att hemokromatos ska misstänkas vid förhöjd järnmättnad, stegrade aminotransferaser utan annan förklaring eller oklara artrosliknande ledbesvär i småleder kan leda till tidig upptäckt och behandling. Diagnostiken i okomplicerade och tidiga fall är enkel, liksom behandlingen, och kan genomföras inom primärvården om det finns tillgång till sjuksköterska med kompetens att göra venesectio. Tidigt insatt behandling förebygger de komplikationer som kan uppkomma vid långt gången sjukdom, framför allt levercirros.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
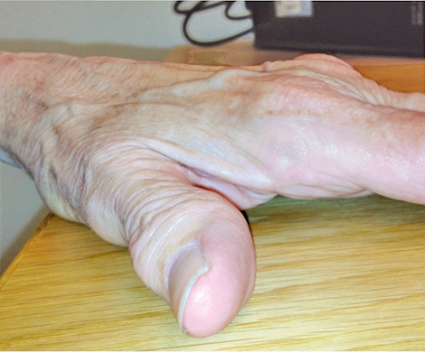
Figur 1. Bild av en hand med typiska uppdrivningar av metakarpofalangealled II och III.
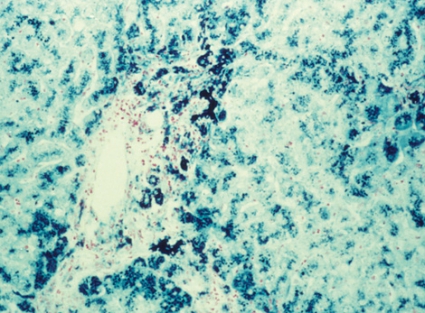
Figur 2. Leverbiopsivävnad som färgats för järn med Perls’ blue. Man ser blå pigmentkorn som är lokaliserade i hepatocyterna med koncentration i de periportala områdena. Pigment ses även i gallgångscellerna.
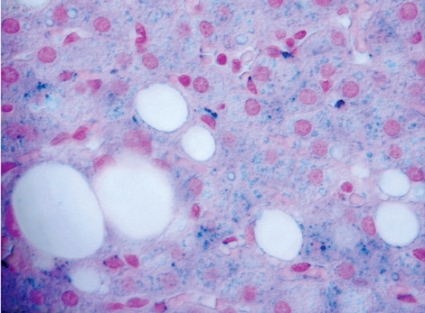
Figur 3. Levervävnad från en patient med metabolt syndrom med högt ferritin. Det föreligger fettinlagring i hepatocyterna (de vita runda områdena). Järnfärgning visar en mer diffus järninlagring än den i Figur 2, med hög koncentration järn i mindre celler, som utgörs av Kupfferceller.
Referenser
1. Aleman S, Endalib S, Stål P, et al. Health check-ups and family screening allow detection of hereditary hemochromatosis with less advanced liver fibrosis and survival comparable with the general population. Scand J Gastroenterol. 2011;46:1118-26.
2. Lainé F, Reymann JM, Morel F, et al. Effects of phlebotomy therapy on cytochrome P450 2e1 activity and oxidative stress markers in dysmetabolic iron overload syndrome: a randomized trial. Aliment Pharmacol Ther. 2006;24:1207-13.
3. Siddique A, Kowdley KV. Review article: the iron overload syndromes. Aliment Pharmacol Ther. 2012;35:876-93.
4. Cardoso EM, Stål P, Hagen K, et al. HFE mutations in patients with hereditary haemochromatosis in Sweden. J Intern Med. 1998;243:203-8.
5. Sun CC, Vaja V, Babitt JL, et al. Targeting the hepcidin-ferroportin axis to develop new treatment strategies for anemia of chronic disease and anemia of inflammation. Am J Hematol. 2012;87:392-400.
6. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393-408.
7. Deugnier Y, Turlin B. Pathology of hepatic iron overload. Semin Liver Dis. 2011;31:260-71.
8. Sirlin CB, Reeder SB. Magnetic resonance imaging quantification of liver iron. Magn Reson Imaging Clin N Am. 2010;18:359-81.
9. Adhoute X, Foucher J, Laharie D, et al. Diagnosis of liver fibrosis using FibroScan and other noninvasive methods in patients with hemochromatosis: a prospective study. Gastroenterol Clin Biol. 2008;32:180-7.
10. Carroll GJ, Breidahl WH, Olynyk JK. Characteristics of the arthropathy described in hereditary hemochromatosis. Arthritis Care Res (Hoboken). 2012;64:9-14.
11. Wood MJ, Powell LW, Dixon JL, et al. Clinical cofactors and hepatic fibrosis in hereditary hemochromatosis: the role of diabetes mellitus. Hepatology. 2012;56(3):904-11.
12. Elmberg M, Hultcrantz R, Ekbom A, et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology. 2003;125:1733-41.
13. Elmberg M, Hultcrantz R, Ebrahim F, et al. Increased mortality risk in patients with phenotypic hereditary hemochromatosis but not in their first-degree relatives. Gastroenterology. 2009;137:1301-9.
14. Rombout-Sestrienkova E, Nieman FH, Essers BA, et al. Erythrocytapheresis versus phlebotomy in the initial treatment of HFE hemochromatosis patients: results from a randomized trial. Transfusion. 2012;52:470-7.
15. Elmberg M, Hultcrantz R, Simard JF, et al. Risk of ischaemic heart disease and cardiomyopathy in patients with haemochromatosis and in their first-degree relatives: a nationwide, population-based study. J Intern Med. 2012;272(1):45-54.