Sammanfattat
I behandlingsstudier behövs en kontrollgrupp för att skilja behandlingseffekten från placeboeffekter och naturalförlopp. Även kontrollgruppen kan få en aktiv behandling.
Randomisering minskar risken för att studiens resultat påverkas av förväxlingsfaktorer.
Inklusions- och exklusionskriterier avgör vilka patienter som får vara med i studien. Valet av kriterier utgör en balansgång mellan generaliserbarhet och statistik styrka.
Kliniska utfallsmått är mer tillförlitliga än surrogatmått, men kan kräva större och mer långvariga studier.
Behandlingsavbrott och andra avvikelser från studieprotokollet kan hanteras på olika sätt när utfallsdata analyseras.
Kliniska läkemedelsstudier delas in i fyra faser och styrs av ett särskilt regelverk.
Kraven på vetenskaplig dokumentation har blivit allt hårdare när nya terapier ska introduceras i vården. Särskilt tydligt är detta inom läkemedelsområdet, där registrering av ett nytt läkemedel ofta föregås av mycket stora och kostsamma randomiserade studier utformade för att styrka den terapeutiska effekten.
De kliniska prövningar som föregår introduktionen av ett nytt läkemedel brukar delas in i tre faser, I–III. I fas I undersöks läkemedlets egenskaper på ett litet antal friska försökspersoner. I fas II fastlår man att terapin överhuvudtaget har en effekt och försöker hitta optimal dosering. I de stora fas III-studierna riktar man in sig på behandlingseffektens storlek och jämförelser med redan etablerade terapier. Studier som genomförs efter att läkemedlet kommit ut på marknaden klassas som fas IV och syftar ofta till att karakterisera läkemedlets biverkningar och säkerhetsprofil [1].
Studier av ickefarmakologiska behandlingar inom till exempel kirurgi och psykoterapi innebär särskilda utmaningar. Tillsammans med färre formella dokumentationskrav har det bidragit till att randomiserade kontrollerade studier här varit mindre vanliga. Exempelvis har det ofta varit svårt att åstadkomma adekvata placebobehandlingar och upprätthålla blindning. Med rätt metoder går problemen trots allt ofta att lösa, och randomiserade studier har blivit ett självklart inslag även inom den ickefarmakologiska behandlingsforskningen.
Målet med denna artikel är att ge en översiktlig beskrivning av metoder och begrepp som är centrala vid vetenskapliga studier av behandlingseffekt och att belysa de särskilda problem man kan ställas inför vid genomförandet av sådana studier.
Kontrollgrupp. När man vill undersöka effekten av en behandling räcker det nästan aldrig med att bara studera patienter som får denna behandling. Ett skäl är att sjukdomssymtomen ofta varierar över tid. Om patienten förbättras efter att behandlingen påbörjats behöver det inte betyda att terapin varit effektiv. Många patienter mår också bättre bara av att vara med i en studie där de följs upp noggrant och får någon slags behandling som de hoppas ska hjälpa dem. Även om denna placeboeffekt är en del av den totala behandlingseffekten, och något man vill dra nytta i den kliniska verkligheten, får den inte räknas in vid registreringen av ett nytt läkemedel. Effektstudierna syftar därför till att specifikt mäta läkemedlets farmakologiska effekter och skala bort eventuella placeboeffekter.
För att kunna mäta enbart de farmakologiska effekterna behöver man jämföra utfallet hos de läkemedelsbehandlade patienterna med utfallet hos en kontrollgrupp. Historiskt har man ofta behandlat patienterna i kontrollgruppen med placebo, en behandling som till det yttre är oskiljbar från den riktiga behandlingen men som saknar den farmakologiskt aktiva komponenten. Den eventuella förbättring man ser hos patienterna i kontrollgruppen återspeglar då placeboeffekter, naturalförlopp och liknande, medan skillnaden i effekt mellan behandlings- och kontrollgruppen anses återspegla läkemedlets farmakologiska effekt [2].
Eftersom det i dag finns verksamma behandlingar mot många av våra vanliga sjukdomar kan det vara etiskt tveksamt att behandla kontrollgruppen med placebo, då det innebär att man undanhåller dem en fungerande behandling. Det har därför blivit vanligare att nya behandlingar inte jämförs med placebo utan med en annan redan etablerad behandlingsform.
När man jämför två aktiva behandlingar på detta sätt är det långtifrån alltid som den nya behandlingen är uppenbart bättre i termer av behandlingseffekt. Tvärtom är många nya terapier likvärdiga med de etablerade, men kan ha andra fördelar som lindrigare biverkningar eller liknande. När man inte räknar med att kunna påvisa någon skillnad i behandlingseffekt mellan grupperna kan man välja att analysera resultaten enligt en »non-inferiority«-modell (inte sämre än). I en sådan analys visar man att eventuella skillnader i effekt mellan behandlingarna är så små att de kan anses kliniskt försumbara enligt definitioner man enats om på förhand (Figur 1). När en behandling jämförs med en annan är det viktigt att kontrollbehandlingen är den bästa tillgängliga, eftersom man annars hamnar i en etisk problematik liknande den med placebo. Jämförelser med en suboptimal behandling ger en överdrivet positiv bild av den nya behandlingen [3].
I vissa fall är det möjligt att låta varje patient i studien vara sin egen kontroll. I sådana »crossover-studier« behandlas varje patient under separata tidsperioder med de terapier man vill jämföra, varefter man jämför behandlingseffekten i de olika perioderna. Fördelen med denna design är att effekten av behandlingarna mäts mot en identisk bakgrund av de patientfaktorer som kan påverka utfallet, vilket gör att skillnaden i utfall mellan grupperna som kan hänföras till annat än att behandlingen minskar. Detta gör det lättare att statistiskt urskilja en verklig behandlingseffekt av en given storlek. Studien kan därför oftast genomföras med färre patienter. För att patienterna ska kunna vara sina egna kontroller krävs dock att de symtom eller utfallsmått man försöker behandla är relativt stabila över tid, och att man låter det gå så lång tid mellan behandlingsomgångarna att effekten av den första behandlingen helt försvunnit innan man påbörjar nästa omgång (så kallad wash out) [4].
Observationella studier. Behandlingseffekter kan utvärderas i observationella studier där man följer upp hur det gått för patienter som ordinerats behandlingen. Detta kan till exempel gå till så att man identifierar kohorter av patienter med samma sjukdom som antingen fått den behandling man är intresserad av (behandlingsgrupp) eller en alternativ behandling (kontrollgrupp) och jämför behandlingsresultaten i de båda grupperna. Fördelen med sådana observationsstudier är att de är kostnadseffektiva och att man genom att utnyttja retrospektiva data kan utvärdera långa behandlingstider utan att ägna år åt datainsamling. Den stora nackdelen är risken för påverkan av förväxlingsfaktorer (confounding), i synnerhet »confounding by indication«. Med detta menas att valet av behandling styrts av patientens diagnos, sjukdomsintensitet eller liknande faktorer som i sin tur påverkar behandlingsutfallet. I detta läge kan det se ut som om behandlingarna har olika effekt trots att det man mätt egentligen är skillnaden i prognos mellan två olika patientpopulationer (Figur 2). Om man känner till vilka förväxlingsfaktorerna är och registrerar dem noga går det att korrigera för dem i den statistiska analysen, men då vissa faktorer inte går att mäta och andra förblir helt okända kan man aldrig helt skydda sig mot confounding.
Trots svårigheterna att med hjälp av observationella data skatta behandlingseffekter erbjuder det svenska sjukvårdssystemets struktur och tillgången till nationella register osedvanligt gynnsamma förutsättningar för denna typ av studier. Dessa möjligheter har ytterligare förbättras med Läkemedelsregistret, som innehåller heltäckande information om uthämtande av receptbelagda läkemedel på individnivå.
Randomiserade studier. I en randomiserad studie fördelas deltagarna slumpmässigt till behandlingar som ska jämföras. Den viktigaste fördelen med att på detta sätt låta slumpen avgöra vilken behandling patienten får är att det minskar risken för förväxlingsfaktorer. Om randomiseringen går rätt till är sannolikheten att hamna i den ena eller andra gruppen helt oberoende av patientens egenskaper, och man hoppas därför slippa en systematisk snedfördelning av patientfaktorer som kan påverka utfallet. Om det enda som skiljer studiegrupperna åt är vilken behandling de fått kan man känna sig betydligt säkrare på att de skillnader man ser i utfall verkligen beror på behandlingen. Randomisering betraktas därför i dag nästan som ett måste vid behandlingsstudier, åtminstone om dessa ska ligga till grund för registreringen av ett nytt läkemedel eller en ny indikation för ett redan registrerat läkemedel.
Även om randomiseringen påtagligt minskar risken för snedfördelning av patientegenskaper som påverkar utfallet måste man vara medveten om att det på inget sätt garanterar en jämn fördelning. Det är fortfarande fullt möjligt att patienterna av en ren slump råkar fördelas så att till exempel alla de äldsta hamnar i en av studiegrupperna och att detta har en högst påtaglig effekt på studieresultatet. För att förebygga sådan slumpvis snedfördelning av faktorer med välkänt stor inverkan på studiens resultat kan man använda sig av stratifierad randomisering. Den går till så att man grupperar patienterna utifrån kön, sjukdomsintensitet eller någon egenskap man vet med sig påverkar utfallet. Sedan randomiserar man grupperna separat på ett sådant sätt att egenskapen i fråga fördelas jämt i de olika studiearmarna (se blockrandomisering nedan). Man kan också, precis som i observationsstudier, statistiskt justera resultaten för olika förväxlingsfaktorer, men bara om dessa är kända och kan mätas.
Det vanliga är att randomisera enskilda patienter till de behandlingar som ska jämföras, men ibland är detta inte möjligt. Om man exempelvis vill undersöka effekten av att införa en ny typ av teamarbete vid öppenvårdsmottagningar ligger det närmare till hands att randomisera hela mottagningar snarare än enskilda patienter. Vid denna typ av grupprandomisering krävs särskilda statistiska analysmetoder, eftersom patienterna vid en mottagning ofta har gemensamma egenskaper som skiljer dem från patienterna vid en annan mottagning, vilket i sig kan påverka behandlingsresultatet. Analysmetoder som tar hänsyn till sådana klustereffekter gör att man oftast måste inkludera fler patienter än om randomiseringen kunnat göras på individnivå [5].
Randomiseringen kan göras på många olika sätt, till exempel med slantsingling, lottdragning eller datorgenerade slumptal. Det viktiga är att utfallet verkligen är slumpmässigt, och det är exempelvis inte tillräckligt att fördela patienterna efter vilken veckodag de kommit till mottagningen, eftersom patienter som söker vård på måndagar mycket väl kan skilja sig systematiskt från dem som dyker upp på fredagar. För att inte utfallet ska påverkas av förväntningar på behandlingen försöker man oftast hemlighålla utfallet av randomiseringen genom blindning. I en dubbelblindad studie är både patienten och den som behandlar omedveten om vilken behandling patienten randomiserats till, men terminologin är inte glasklar. Om exempelvis effekten utvärderas av en annan person än den som behandlat patienten finns det ju tre parter inblandade som alla medvetet eller omedvetet kan påverka studieresultatet om de känner till utfallet av randomiseringen.
Det har hänt att den som behandlar patienten manipulerat randomiseringen för att se till att särskilt ömmande fall får aktiv behandling och inte placebo. Även om detta vanligen görs av omsorg om den enskilda patienten innebär manipuleringen att studieresultatens validitet äventyras. Detta kan vara till skada för framtida patienter, vars terapival baseras på en felaktigt skattad behandlingseffekt. Bland annat för att undvika denna typ av snedfördelning väljer man ofta att administrera randomiseringen centralt utan möjlighet för behandlaren att påverka utfallet.
Vid helt slumpmässig randomisering finns en risk att grupperna blir olika stora, särskilt om studien är liten. Detta försämrar den statistiska styrkan, och för att försäkra sig om en jämn fördelning kan man utnyttja blockrandomisering. Vid blockrandomisering slumpas inte patienterna var för sig utan i mindre block om till exempel fyra patienter. Om man vill ha lika stora grupper utformas blocken så att två av patienterna styrs till den ena behandlingsgruppen och två till den andra (den exakta ordningsföljden inom blocket är slumpmässig och det som egentligen avgörs av randomiseringen). Detta innebär att av de fyra patienter som först inkluderas i studien måste två hamna i vardera studiegruppen, av de fyra som inkluderas därnäst hamnar åter två i vardera gruppen och så vidare. På så sätt har man kontinuerligt ett lika stort patientinflöde i båda grupperna, vilket inte alls behöver vara fallet vid randomisering på individnivå (Figur 3) [6].
Oavsett randomiseringsmetod är målet oftast att få lika stora grupper, eftersom den statistiska styrkan (förmågan att påvisa en behandlingseffekt av en viss storlek) då blir störst. Av etiska eller ekonomiska skäl kan man ibland vilja minimera antalet patienter i en av grupperna. Det är då möjligt att skapa olika stora grupper. Det är sällan någon poäng med större obalanser än 1:3, eftersom ytterligare patienter i den större gruppen tillför mycket lite i termer av statistisk styrka.
Patientselektion. Vilka patienter som får vara med i studien bestäms med hjälp av inklusions- och exklusionskriterier. Dessa kriterier kan exempelvis specificera vilka åldersintervall patienterna får tillhöra, vilka sjukdomskriterier som måste vara uppfyllda eller att patienterna inte får ta några andra läkemedel som påverkar den sjukdom man vill behandla eller som interagerar med den behandling man vill studera. Om patienterna uppvisar en stor variation i fråga om olika individegenskaper som i sin tur påverkar den sjukdom eller de symtom man vill studera bidrar detta till ett statistiskt brus (variationer i utfall som är orelaterade till behandlingen) mot vilket effekten av den studerade behandlingen blir svårare att urskilja.
Som ett hypotetiskt exempel kan man tänka sig ett läkemedel som förlänger överlevnaden vid en specifik tumörsjukdom med två månader. I en mycket homogen patientpopulation där alla patienter utan behandling överlever mellan tre och fyra månader blir effekten lätt att urskilja eftersom patienterna plötsligt överlever fem till sex månader, längre än någon skulle ha gjort utan behandling. I en mer heterogen patientgrupp med olika avancerad tumörsjukdom, åldrar, samsjuklighet etc varierar överlevnaden hos obehandlade mellan noll och tolv månader. I denna grupp blir samma läkemedelseffekt mycket svårare att urskilja eftersom den drunknar i den stora spontana variationen i överlevnad.
Svårigheterna att urskilja effekten brukar beskrivas som minskad statistisk styrka och innebär att fler patienter måste inkluderas för att resultaten ska bli användbara. Då stora studier är dyra att genomföra kan det vara praktiskt att genom inklusions- och exklusionskriterierna välja ut en väldefinierad homogen grupp av patienter där behandlingseffekten kan urskiljas i ett mindre patientmaterial. Problemen med detta angreppssätt är att de hårt sållade studiedeltagarna kan bli mycket olika den brokiga skara av patienter som i framtiden ska behandlas med läkemedlet. Studiedeltagare som inte är representativa för den verkliga patientpopulationen ger studien låg »extern validitet« och väcker frågor om hur giltiga resultaten är för de patienter man dagligen möter inom sjukvården.
När man mäter effekten på relativt ovanliga utfall som död eller slaganfall kan man hålla nere studiestorleken genom att enbart inkludera patienter med extra hög risk för dessa utfall. Även här kan man få problem med den externa validiteten eftersom resultaten inte okritiskt går att applicera på patienter med lindrigare sjukdom eller färre riskfaktorer.
Utfallsmått. När man studerar behandlingseffekter är det givetvis av central betydelse hur utfallet mäts. Målet med terapin är att åstadkomma något slags klinisk effekt, till exempel att lindra symtom, ge fullständig utläkning eller att förebygga komplikationer. Det bästa är naturligtvis om det går att mäta effekten på just dessa kliniska mål. Om det kliniska utfallet är sällsynt kan det dock krävas mycket stora studier för att påvisa effekten. Inte minst förebyggande behandlingar kan behöva studeras under långa tider för att deras effekter på kliniska utfall ska kunna styrkas.
Då stora och långvariga studier är mycket dyra att genomföra väljer man ofta att i stället studera så kallade surrogatutfall som i sig är ointressanta för patienten men som man av erfarenhet vet är associerade med de kliniska utfall man hoppas påverka. Det är exempelvis relativt enkelt att visa att en ny blodtryckssänkare sänker blodtrycket och utifrån detta dra slutsatsen att läkemedlet på lång sikt kommer att minska risken för slaganfall och död hos hypertoniker.
Problemet med surrogatutfall är att man inte kan vara helt säker på hur det verkliga sambandet mellan surrogatutfallet och det kliniska utfallet ser ut. Olika typer av blodtryckssänkare kan till exempel ha olika effekter på mortaliteten även om de ger identisk blodtryckssänkning, eftersom de också kan ha kliniskt viktiga effekter som medieras via andra mekanismer än just blodtryckssänkningen. På samma sätt kan statinernas effekter på blodfetterna inte fullt ut förutsäga den kliniska nyttan om deras antiinflammatoriska egenskaper också påverkar prognosen. Tolkningen av behandlingseffekter som enbarts mätts i form av surrogatutfall kräver extra stor försiktighet och en beredskap för att framtida studier på kliniska utfall kan bjuda på otrevliga överraskningar om surrogatmåttets träffsäkerhet överskattats.
Objektiva utfall som till exempel död kan ofta mätas med större säkerhet än mått med ett större inslag av subjektivitet som exempelvis smärta eller livskvalitet. Av detta skäl strävar man ofta efter att hitta objektiva effektmått, men även om det med stor säkerhet går att avgöra om patienten överlevt eller dött under den tid studien pågått får man inte glömma att själva dödsorsaken ofta är svår att säkerställa.
Avvikelser från studieprotokollet. Det är inte säkert att man har möjlighet att följa upp alla patienter som inkluderats i en studie, eftersom vissa avbryter deltagandet i förtid. Sådana patientbortfall kan bero på att patienten flyttar, att sjukdomar som omöjliggör deltagande tillstöter eller att patienten helt enkelt inte längre vill vara med. Ett annat potentiellt problem är patienter som visserligen stannar kvar i studien, men som inte tar den ordinerade behandlingen på det sätt det var tänkt.
Sådana bortfall och avvikelser från studieprotokollet måste på något sätt hanteras när resultaten från studien analyseras. Ett sätt är att enbart analysera utfallet hos de patienter som fullföljt studien på ett korrekt sätt. En nackdel med en sådan per protokoll-analys (PP) är att den bortser från att bortfall och följsamhet till studieprotokollet i sig kan ha varit relaterade till behandlingsutfallet, vilket ger en snedvriden bild av terapins effekter. Om patienterna till exempel avbrutit behandlingen eller hoppat av studien på grund av bristande effekt ger analysen en överskattning av behandlingseffekten, eftersom de patienter som fullföljt studien är just de som haft god effekt.
Analys per protokoll innebär dessutom att man frångår de randomiserade grupper som skapats för att ge en jämn fördelning av kända och okända patientegenskaper. Eftersom det inte är slumpen som avgör vilka patienter som exkluderas i analysen finns en uppenbar risk att patientfaktorer av betydelse för behandlingsutfallet blir snedfördelade, vilket kan leda till confounding. Då det inte alltid är uppenbart vilka avvikelser som är tillräckligt allvarliga för att patienten ska exkluderas kommer per protokoll-analysen också att föra in ett nytt element av subjektivitet när studieledningen tvingas ta ställning till vilka patienter som ska få ingå i dataanalysen. Detta är olyckligt, då man med randomisering, blindning och liknande strävat efter att minska inflytandet av just sådana subjektiva beslut.
Ett alternativt sätt att analysera utfallet är enligt intention to treat (ITT) [7]. I denna analys inkluderas alla patienter som påbörjat studien och man räknar med att de fått den behandling de randomiserats till oavsett hur följsamheten sett ut eller om de senare avbrutit deltagandet (Figur 4). Fördelen med en sådan analys är att den bevarar randomiseringens slumpvisa gruppindelning och förhindrar att systematiska bortfall eller exklusioner till följd av protokollavvikelser introducerar nya snedfördelningar som påverkar studieresultatet. Den uppenbara nackdelen är att studiegrupperna blandas upp med individer som inte alls fått den avsedda behandlingen, vilket ger en utspädningseffekt och en underskattning av effekten hos patienter som verkligen exponerats för terapin.
Intention to treat-analys ger vanligen en mer konservativ bild av behandlingseffekten och är den analysmetod som i första hand brukar efterfrågas av läkemedelsmyndigheter och vetenskapliga tidskrifter. Ännu mer informativt är att presentera resultaten enligt både intention to treat- och per protokoll-analys och att noga fundera över eventuella skillnader i utfall mellan de två analyserna.
Ett problem vid intention to treat-analys kan vara att man saknar utfallsdata för patienter som avbrutit deltagandet i förtid eller där man av andra skäl saknar utfallsdata. I detta läge kan man bli tvungen att göra ett antagande om hur det gått för patienterna. Det finns att stort antal mer eller mindre komplicerade algoritmer för att tilldela (imputera) patienterna ett fiktivt utfall i de fall uppgiften saknas [8]. Även om metodernas träffsäkerhet varierar från situation till situation blir slutresultatet oftast mer tillförlitligt än om patienterna helt enkelt exkluderats som i en per protokoll-analys.
God klinisk praxis. Avslutningsvis kan nämnas att alla kliniska prövningar, det vill säga prospektiva studier där läkemedel ges till människor i syfte att studera behandlingens effekter, som genomförs inom EU, USA eller Japan regleras av ett särskilt regelverk utformat av samarbetsorganet International Conference on Harmonisation (ICH). Riktlinjerna, som går under namnet Good Clinical Practice (god klinisk praxis), ger detaljerande anvisningar om bland annat hur prövarnas inbördes ansvar fördelas, hur studiens design och resultat ska dokumenteras och hur studiedeltagarnas integritet och säkerhet ska säkerställas [1].
Det är inte enbart datainsamlingen och de kliniska delarna av studien som måste dokumenteras på rätt sätt, utan även den avslutande dataanalysen. Vilka utfall som ska studeras och vilka statistiska metoder som ska användas måste beslutas och dokumenteras redan i ett tidigt skede eftersom det i efterhand är alltför lätt att manipulera utfallet genom att prova sig fram till en analys som ger just det resultat man hoppats på. Sådana »multipla test« kan också åstadkommas om man delar in patientmaterialet i olika subgrupper som analyseras var för sig. Även här ökar risken påtagligt att man ska hitta signifikanta behandlingseffekter också där sådana inte finns.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
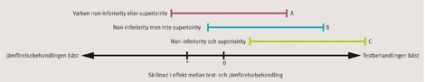
Figur 1. Behandling A, B och C jämförs med en jämförelsebehandling. Den fördefinierade non-inferioritygränsen (*) motsvarar den minsta effektskillnad som anses kliniskt betydelsefull. Konfidensintervallet för A sträcker sig förbi non-inferiority-gränsen. Non-feriority-kravet är inte uppfyllt. Konfidensintervallet för B ligger till höger om non-inferiorityg-ränsen och non-inferiority-kravet är därför uppfyllt. Då intervallet ligger till höger om 0 (behandlingarna likvärdiga) är C signifikant bättre än jämförelsebehandlingen.
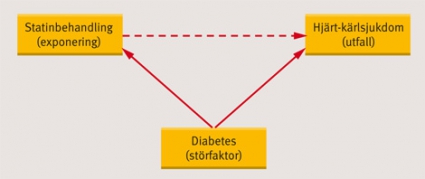
Figur 2. Exempel på förväxlingsfaktorer. Indikationen för statinbehandling av hyperlipidemi anses extra stark hos diabetiker eftersom dessa har en ökad risk för hjärt–kärlsjukdom. Då diabetes ökar sannolikheten både för exponering och utfall uppstår ett skensamband där statiner tycks öka risken för kardiovaskulär sjukdom. I en observationell studie utan korrektion för förväxlingsfaktorer mäter man den sammanlagda effekten av statinbehandlingen (gynnsam) och olika förväxlingsfaktorer (gynnsamma eller ogynnsamma).
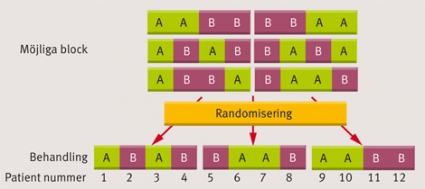
Figur 3. Blockrandomisering. Patieterna randomiseras i block om till exempel fyra efter hand som de inkluderas. Inom blocken får två patienter alltid behandling A och två patienter behandling B. På så sätt garanteras en jämn fördelning mellan grupperna.
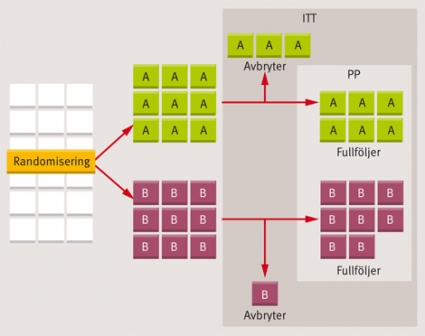
Figur 4. Per protokoll- och intention to treat-analys. I exemplet randomiseras nio patienter till behandling med A respektive behandling B. Under studiens gång avbryter tre patienter behandling A och en behandling B. Vid analys per protokoll (PP) används bara data från dem som fullföljt studien (blå streckad linje). Vid analys enligt intention to treat (ITT) inkluderas alla randomiserade patienter (grön streckad linje).
(uppdaterad 2021-09-01)
Referenser
1. Lemne C. Handbok för kliniska prövare. 4th ed. Lund: Studentlitteratur; 2002.
2. Ernst E. Placebo: new insights into an old enigma. Drug Discov Today. 2007;12:413-8.
3. Snapinn, SM. Noninferiority trials. Curr Control Trials Cardiovasc Med. 2000;1:19-21.
4. Sibbald B, Roberts C. Understanding controlled trials. Crossover trials. BMJ. 1998;316:1719.
5. Christie J, O’Halloran P, Stevenson M. Planning a cluster randomized controlled trial: methodological issues. Nurs Res. 2009;58:128-34.
6. Pandis N. Randomization. Part 1: sequence generation. Am J Orthod Dentofacial Orthop. 2011;140:747-8.
7. Montori VM, Guyatt GH. Intention-to-treat principle. CMAJ. 2001;165:1339-41.
8. Olsen MK, Stechuchak KM, Edinger JD, et al. Move over LOCF: principled methods for handling missing data in sleep disorder trials. Sleep Med. 2012;13:123-32.
Summary
In studies of treatment effect, a control group is necessary to differentiate therapeutic effect from placebo effects and the natural course of the disease. The control group could receive an alternative therapy or placebo, and the risk of confounding is reduced by randomized study group allocation. Inclusion and exclusion criteria define which patients may participate in the study. The choice of these criteria is often a trade-off between statistical power and external validity. Clinical outcome measures may be more relevant compared to surrogate endpoints, but usually requires larger studies with longer duration. An intention-to-treat analysis includes all randomized patients, while protocol a per-protocol analysis excludes drop-outs and patients violating the study protocol. Clinical trials are divided into four phases and are governed by the Good Clinical Practice (GCP) guidelines.
Jonatan Lindh
Correspondence: Jonatan Lindh, Avdelningen för klinisk farmakologi, Karolinska universitetssjuk-huset, Huddinge, SE-141 86 Stockholm, Sweden
jonatan.lindh@ki.se