Hereditärt hyperferritinemi–kataraktsyndrom är en sällsynt autosomalt dominant sjukdom med okänd prevalens.
Sjukdomen beror på mutationer i genen för L-ferritin och medför okontrollerad tillverkning av L-ferritin utan att det finns ett järnöverskott.
Den enda sjukdomsmanifestation som finns beskriven är bilateral katarakt beroende på inlagring av ferritinkomplex i linserna. Katarakten ger oftast symtom före 30 års ålder.
Ingen annan behandling än kataraktoperation är nödvändig och patienten ska inte venesectiobehandlas.
Det kan vara av värde att läkare som utreder ferritinstegring och ögonläkare känner till sjukdomen.
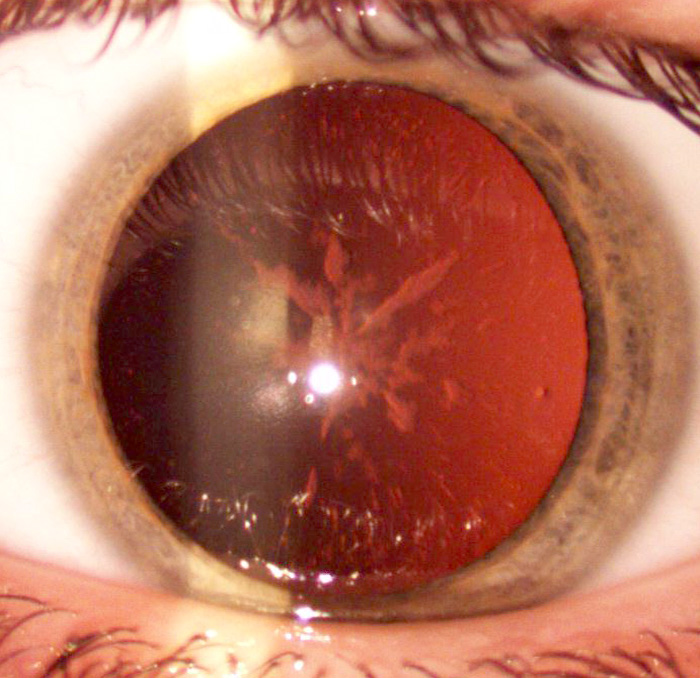
Figur 1. Lins hos patientens 17-årige son. Central katarakt med starrekrar och mindre infiltrat i periferin.
Hereditärt hyperferritinemi–kataraktsyndrom (HHCS) är en sällsynt autosomalt dominant sjukdom. Sjukdomen beskrevs första gången 1995 när man upptäckte familjer med oförklarlig hyperferritinemi och tidigt insättande katarakt [1, 2]. Den bakomliggande sjukdomsmekanismen med mutationer i genen för L-ferritin upptäcktes strax efteråt [3, 4]. Sjukdomen fick så småningom namnet hereditärt hyperferritinemi–kataraktsyndrom (HHCS).
Prevalens och patogenes
Prevalensen är inte känd. I en italiensk screeningundersökning av drygt 3 000 blodgivare och knappt 13 000 patienter med katarakt hittades inga patienter med de kända sjukdomsmutationerna [5]. I en genomgång av patienter med katarakt från södra Australien uppskattades prevalensen till 1/200 000, men denna siffra bedömdes som för låg eftersom sjukdomen är dåligt känd av oftalomologer och ferritin inte mättes rutinmässigt [6].
Ferritin är en makromolekyl som består av 24 subenheter av H- och L-ferritin, som tillsammans bildar ett skal kring en hålighet, där upp till 4 500 järnatomer kan lagras säkert. Bildandet av ferritin i cellen styrs av järnreglerande proteiner (IRP) och en gensekvens på kromosom 19 kallad IRE (iron responsive element). Vid HHCS har en mutation förändrat strukturen på IRE, vilket medför en okontrollerad syntes av L-ferritin. Denna syntes sker oberoende av cellens järninnehåll. För närvarande finns 31 olika mutationer beskrivna [7].
(Järnregleringen i cellen har förenklats starkt här. En mer fullständig förklaring finns i en översiktsartikel [8].)
Klinisk bild
HHCS karaktäriseras således av överproduktion av L-ferritin utan att det finns ett järnöverskott. Så vad medför det i praktiken?
L-ferritin ansamlas i kroppsvävnader men verkar vara icke-
funktionellt och medför inga beskrivna organskador. Undantaget är ögonens linser. Här sker en gradvis inlagring av ferritinkomplex, som ger katarakt. Katarakten beskrivs som central, ofta med radierande starrekrar (Figur 1). Mindre infiltrat ses också perifert i linsen, och vid progress fylls det mellanperifera området ut. Vid mikroskopisk undersökning ses små vita »brödsmuleliknande« infiltrat som kan konfluera till större kristalliknande utfällningar. Ibland kan man också se små vakuoler. S-ferritinnivån verkar inte korrelera med graden av katarakt. Katarakten ger oftast symtom före 30-årsåldern, i många fall redan före 10 års ålder.
För övrigt är patienten opåverkad, har normala blodprov och normal järnmättnad. Differentialdiagnoser kan vara hemokromatos, inflammation, malignitet och leverpåverkan/alkoholmissbruk. Särskilt hemokromatos är värd att nämna. Det finns många beskrivningar av patienter med HHCS som felaktigt fått diagnosen hemokromatos och utsatts för venesectio. Denna behandling har ingen effekt, och patienten utvecklar snabbt järnbristanemi.
Fallet
Fallet rör en 55-årig kvinna, tidigare väsentligen frisk. Hon sökte på vårdcentral för problem med upprepad aftös stomatit. Blodprov togs, bl a S-ferritin, som var förhöjt till 1 100 μg/l (referensintervall 20–150). Järnmättnaden var normal, 26 procent. Remiss skrevs till hematolog på misstanke om hemokromatos. Vid ett nytt besök var patienten välmående.
Förnyad kontroll av S-ferritin och järnmättnad visade oförändrade värden. Utredning påbörjades. Hemokromatosgenotypning visade att patienten var heterozygot för H63D. Blodprov i övrigt var normala: Hb 135 g/l, LPK 6,4 × 109/l, TPK 238 × 109/l. B-celler visade normalfördelning. SR 2 mm. CRP var 3 mg/l, ALAT 0,9 μkat/l, LD 3,1 μkat/l, ALP 1,3 μkat/l, bilirubin 7 μmol/l och PK 1,0 INR. Natrium, kalium, kalcium, albumin och kreatinin var normala. S-proteinanalys (S-elfores) var normal utan inflammation eller påverkan på immunglobuliner. Ett lågt antitrypsinvärde upptäcktes (0,63 g/l), och man misstänkte genetisk antitrypsinbrist. En subklinisk hypotyreos upptäcktes med värdena TSH 12,0 mIE/l, fritt T4 11 pmol/l och TPO-antikroppar 73 kIE/l (referensintervall 0–49). Levotyroxinbehandling startades.
Patienten hade inga sjukdomstecken, och ingen misstanke om alkoholmissbruk förelåg. Ultraljud buk visade en normalstor lever utan patologi. Leverbiopsi gjordes, som visade normal histologisk bild och negativ järnfärgning. Bedömningen blev oklar S-ferritinstegring utan tecken till patologisk järninlagring, leversjukdom, malignitet eller inflammation.
Patienten kontrollerades under två år med glesa intervall. Hon blev eutyreoid av levotyroxinbehandlingen, men ferritinvärdet påverkades inte. Venesectiobehandling inleddes inte. S-ferritin låg stabilt runt 1 200 μg/l, och järnmättnaden var hela tiden normal liksom rutinprov i övrigt. Patienten var fortsatt symtomfri. Vi upplevde det som störande att inte ha en förklaring till ferritinvärdet, och patienten var också undrande över diagnosen och kontrollerna.
En fördjupad anamnesupptagning gjordes. Patienten hade kontrollerats för katarakt i ungdomen och var bilateralt kataraktopererad vid 42 års ålder. En son i övre tonåren kontrollerades för katarakt, och det fanns ytterligare fall av katarakt i släkten. Efter en litteraturgenomgång uppkom stark misstanke om HHCS. Diagnosen bekräftas med molekylärgenetisk analys av IRE-regionen på kromosom 19, där patienten hade mutationen 43G>A. Patientens son har samma avvikelse. Just denna mutation finns tidigare bara beskriven hos en släkt i USA [9].
Diskussion
Hur ska man utreda en patient där man misstänker HHCS? Patienterna presenterar sig med en S-ferritinstegring på ca 1 000–3 000 μg/l, och utredningen får inriktas på att hitta orsakerna. Hereditär hemokromatos ger oftast höga levervärden och hög järnmättnad, över 60 procent. Symtomen vid hemokromatos är trötthet, ledbesvär och leverpåverkan. Mindre vanligt är hjärtpåverkan, diabetes samt gonad- och hypofysinsufficiens. Katarakt finns inte beskriven.
Hemokromatosgenotypning ger nästan alltid diagnosen. I Sverige är 90 procent av patienterna med hereditär hemokromatos homozygota för C282Y, och 5 procent är blandheterozygota för C282Y/H63D. Leverbiopsi för att påvisa ökad mängd järn är inte nödvändig för diagnosen men rekommenderas vid S-ferritin över 1 000 μg/l eftersom det vid den nivån finns risk för levercirros [10]. Om leverbiopsi är kontraindicerad kan järninnehållet i levern uppskattas med MR-undersökning. Övriga orsaker till S-ferritinstegring, som inflammation, malignitet, leversjukdom och alkoholmissbruk, diagnostiseras oftast lätt med provtagning och anamnes.
En patient med HHCS ska ha en isolerad S-ferritinstegring. Alla andra prov ska vara normala och patienten symtomfri. Om andra orsaker till S-ferritinstegring uteslutits och patienten har en familjehistoria med katarakt i unga år är vår uppfattning att man kan nöja sig med detta. Vid tveksamhet kan molekylärgenetisk undersökning göras.
Konklusion
Sjukdomen är benign till sin karaktär. Det kan vara av värde för läkare som utreder förhöjt S-ferritin och ögonläkare att känna till sjukdomen – detta för att patienten ska få besked om tillståndets godartade karaktär och inte gå på kontroller med en okänd diagnos. Det är också viktigt att patienten inte utsätts för venesectiobehandling samt onödiga och potentiellt farliga undersökningar (som leverbiopsi).
Referenser
- Bonneau D, Winter-Fuseau I, Loiseau MN, et al. Bilateral cataract and high serum ferritin: a new dom-inant genetic disorder? J Med Genet. 1995;32:778-9.
- Girelli D, Olivieri O, De Franceschi L, et al. A linkage between hereditary hyperferritinaemia not related to iron overload and autosomal dominant congenital cataract. Br J Haematol. 1995;90:931-4.
- Beaumont C, Leneuve P, Devaux I, et al. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet. 1995;11:444-6.
- Girelli D, Corrocher R, Bisceglia L, et al. Molecular basis for the recently described hereditary hyperferritinemia–cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the »Verona mutation«). Blood. 1995;86:405-3.
- Bozzini C, Galbiati S, Tinazzi E, et al. Prevalence of hereditary hyperferritinemia–cataract syndrome in blood donors and patients with cataract. Haematologica. 2003;88:219-20.
- Craig JE, Clark JB, McLeod JL, et al. Hereditary hyperferritinemia-cataract syndrome: prevalence, lens morphology, spectrum of mutations, and clinical presentations. Arch Ophthalmol. 2003;121:1753-61.
- Millonig G, Muckenthaler M, Muel-ler S. Hyperferritinaemia–cataract syndrome: worldwide mutations and phenotype of an in-creasingly diagnosed genetic disorder. Hum Genomics. 2010;4(4):250-62.
- Cazzola M. Hereditary hyperferritinaemia/cataract syndrome. Best Pract Res Clin Haematol. 2002;15(2):385-98.
- Phillips JD, Warby CA, Kushner JP. Identification of a novel mutation in the L-ferritin IRE leading to hereditary hyperferritinemia–cataract syndrome. Am J Med Genet. 2005;134A:77-9.
- Stål P, Hultcrantz R. Hereditär hemokromatos – en vanlig genetisk sjukdom. Läkartidningen. 2012;46:2097-9.
Summary
Hereditary hyperferritinemia–cataract syndrome (HHCS) is a rare autosomal dominant disease. HHCS is characterized by early onset cataract and high levels of serum ferritin without iron overload. The syndrome is caused by mutations within the iron responsive element segment of the gene for L-ferritin on chromosome 19. Uncontrolled production of L-ferritin leads to deposits of ferritin in the lens of the eye causing cataract, but no other disease symptoms are known. We describe a patient with HHCS and a mutation only reported once before, 43G>A.