Sammanfattat
Creutzfeldt–Jakobs sjukdom är en prionsjukdom som kännetecknas av snabbt progredierande neurodegeneration, som leder till döden inom månader till något år. Eftersom sjukdomen är överförbar är den sedan 1998 anmälningspliktig enligt Smittskyddslagen.
Den heterogena symtombilden, särskilt tidigt i sjukdomsförloppet, gör utredningen av misstänkta fall svår. Säkra diagnostiska metoder för att bekräfta eller utesluta sjukdomen skulle vara av stort värde.
Rutinmässigt innefattar utredningen idag ofta klinisk undersökning, elektroencefalografi, datortomografi eller magnetisk resonanstomografi av hjärnan samt likvoranalys för påvisande av 14-3-3-protein. Definitiv diagnos kan endast ställas neuropatologiskt.
Nya studier talar för att koncentrationsbestämning av total-tau (T-tau) och hyperfosforylerat tau (P-tau) i likvor kan vara komplement till detta utredningsbatteri.
Likvoranalys av T-tau och P-tau kan ha avgörande betydelse i den kliniska diagnostiken av misstänkt Creutzfeldt–Jakobs sjukdom, vilket i artikeln illustreras av några fallbeskrivningar.
Prionsjukdomar är ett samlingsbegrepp för en grupp ovanliga neurodegenerativa sjukdomar med typiska tvättsvampslika (spongiforma) hjärnförändringar, som orsakas av utfällning av ett abnormt veckat endogent protein i hjärnvävnad. Proteinet benämns PrPSc (prion protein scrapie), eftersom det först identifierades i hjärnor från får med scrapie. PrPSc är en förändrad variant av det cellulära proteinet PrPC. Såväl PrPC som PrPSc definieras av samma gen (PRNP) på kromosom 20 och har som regel samma gen- och aminosyrasekvens, med undantag av mycket sällsynta ärftliga former av prionsjukdomar. Det är endast proteinets veckning, dess tertiärstruktur, som skiljer sig åt, och denna skillnad gör PrPSc extremt värmestabilt, resistent mot proteaser och mycket benäget att aggregera [1]. PrPSc kan dessutom konvertera normalt PrPC till felaktigt veckat PrPSc i en kaskadliknande reaktion, vilket är den molekylära bakgrunden till proteinets infektiösa egenskaper.
Prionsjukdomarna innefattar hos människa Creutzfeldt– Jakobs sjukdom, kuru, Gerstmann–Sträussler–Scheinkers sjuk
dom och fatal familjär insomnia [2]. Scrapie drabbar får, och bovin spongiform encefalopati (BSE) drabbar kor.
Creutzfeldt–Jakobs sjukdom, som är den vanligaste prionsjukdomen hos människa, förekommer i fyra varianter:
en mycket ovanlig hereditär form orsakad av mutationer i PRNP-genen,
en iatrogen form överförd via tex forna tiders tillväxthormonpreparat, dura mater- eller hornhinnetransplantat,
en sporadisk form där orsaken är okänd samt
en »ny variant«.
Den sistnämnda har drabbat yngre människor främst i Storbritannien och är sannolikt överförd från BSE-sjuka kor.
Den sporadiska formen är den vanligaste. Det är en åldersrelaterad sjukdom med en ungefärlig årlig incidens av 1,2 fall per miljon invånare i Sverige [3, 4], vilket är i överensstämmelse med övriga världen [5]. Orsaken till varför vissa människor drabbas av sjukdomen är okänd. Mest sannolikt förefaller vara att endogent PrPC på något vis förändras till PrPSc och att detta över en viss tröskelnivå startar den kaskadliknande reaktion som ger upphov till neurodegenerationen.
Utredning
Utredningen av misstänkta fall baseras ofta på en kombination av kliniska, neurofysiologiska och neuroradiologiska kriterier samt likvordiagnostik och neuropatologisk undersökning.
Sjukdomsbilden karakteriseras i typiska fall av snabb demensutveckling tillsammans med multifokala neurologiska symtom, såsom myoklonier, cerebellär ataxi, synstörningar, pyramidala och extrapyramidala symtom samt akinetisk mutism. Datortomografi av hjärnan visar vanligen normala fynd eller ospecifik atrofi. Magnetisk resonanstomografi av hjärnan kan avslöja hyperintensitet i basala ganglierna, och elektroencefalografi (EEG) kan visa på periodiska skarpa vågkomplex, ofta med trifasisk morfologi, men dessa förändringar ses endast hos cirka två tredjedelar av fallen [6, 7]. Albuminkvot och isoelektrisk fokusering av serum och likvor är vanligen normala, liksom celltal i likvor [8].
Med ovanstående kombination kan många viktiga differentialdiagnoser som cerebrovaskulära, infektiösa och inflammatoriska CNS-sjukdomar uteslutas, men snabbt progredierande Alzheimers sjukdom och andra demenssjukdomar kan vara svåra differentialdiagnoser. Diagnostiska kriterier för sporadisk Creutzfeldt–Jakobs sjukdom har utvecklats och möjliggör en indelning av fallen i möjliga, sannolika och säkra (Fakta) [9].
»Hjärnspecifika« proteiner som markörer. I avsaknad av prionspecifik pre mortem-diagnostik har olika »hjärnspecifika« proteiner föreslagits som markörer för Creutzfeldt–Jakobs sjukdom. Gemensamt för samtliga är att de är normalt förekommande neuronala proteiner, som läcker ut till likvor vid Creutzfeldt–Jakobs sjukdom, men även vid andra typer av neuronalt sönderfall. Mest etablerad är kvalitativ analys (immunblot-teknik) av 14-3-3-proteinet i likvor, som i ett flertal studier visat hög (>90 procent) sensitivitet och specificitet för Creutzfeldt–Jakobs sjukdom [10-12]. Falskt positiva fynd har dock setts vid exempelvis encefaliter, stroke, intoxikationer och metastaser [13-16], varför analysen inte lämpar sig för screening av oselekterade patienter med neurologiska symtom. Analys av 14-3-3 utförs vid virusavdelningen på Smittskyddsinstitutet i Solna.
Tau är ett annat neuronspecifikt protein, vars koncentration i likvor kan vara kraftigt förhöjd vid Creutzfeldt–Jakobs sjukdom [17]. Tau förekommer i 6 olika isoformer och kan även fosforyleras, varför en ELISA-metod som känner igen alla isoformerna oberoende av fosforylering har utvecklats (total tau; T-tau) [18]. Flera studier, som tillsammans inkluderar mer än 250 patienter med Creutzfeldt–Jakobs sjukdom, visar att T-tau-koncentration i likvor >1300 ng/l eller >1500 ng/l (beroende på studie) uppvisar en med 14-3-3-analys jämförbar sensitivitet och specificitet för sjukdomen [11, 17, 19, 20].
Koncentrationen av T-tau är dock måttligt till kraftigt förhöjd även vid Alzheimers sjukdom och andra tillstånd med uttalat neuronalt sönderfall, såsom stroke och encefalit [21]. Detta innebär ett möjligt specificitetsproblem, vilket Riemenschneider och medarbetare nyligen angripit i en studie [22]. Vid Alzheimers sjukdom sker en hyperfosforylering av tau, varför inte bara T-tau utan även hyperfosforylerat tau (P-tau) visar ökade koncentrationer i likvor [23]. Tau fosforylerat på treonin 181 kan mätas med en ELISA-metod [24]. Genom att ta en kvot mellan P-tau och T-tau kunde Riemenschneider och medarbetare helt separera patienter med Creutzfeldt–Jakobs sjukdom från patienter med lindrig och måttlig/svår Alzheimers sjukdom, patienter med frontallobsdemens och friska kontroller [22].
I Figur 1 har vi räknat om deras resultat från pmol/l till ng/l (som vi använder vid laboratoriet för klinisk kemi, sektionen för neurokemi, Sahlgrenska Universitetssjukhuset/Mölndal) och beräknat T-tau/P-tau-kvoter (notera att vi inverterat kvoten för att göra resultaten mer överskådliga). I Riemenschneiders och medarbetares studie hade alla 20 patienter med Creutzfeldt–Jakobs sjukdom T-tau/P-tau-kvot över 78 (medelvärde 169), medan det högsta värdet hos kontroller och patienter med andra demenssjukdomar var 12,8 [22].
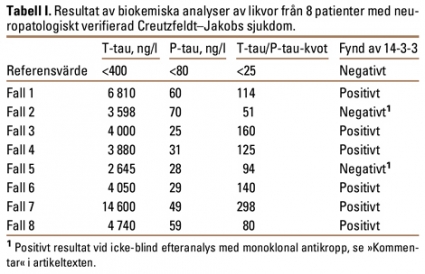
Nedan beskrivs fyra fall som illustrerar den heterogena kliniska bilden samt användbarheten av T-tau/P-tau-kvoten i likvor vid misstänkt Creutzfeldt–Jakobs sjukdom. Vidare rapporterar vi resultaten från ytterligare fyra patienter (fall 5–8) med neuropatologiskt säkerställd sjukdom (Tabell I).
Fallbeskrivningar
Fall 1. En 65-årig nyligen pensionerad kvinna, som 15 år tidigare genomgått hornhinnetransplantation, insjuknade med trötthet, synproblem, diffust ont i kroppen och viktnedgång. Några månader senare och en vecka före ankomsten till sjukhus drabbades patienten av overklighetskänsla i vänster arm, vilken hon inte längre kunde styra. Detta fenomen går under benämningen »alien hand syndrome« och har tidigare beskrivits i samband med Creutzfeldt–Jakobs sjukdom [25], symtomet kan också ses vid andra neurologiska tillstånd [26].
Vid inläggningen på neurologisk klinik noterades lätt pares och dystaxi i vänster arm, kortikal känselstörning i vänster hand samt intermittent atetos och dystona rörelser i vänster arm. Patienten var lätt kognitivt förlångsammad och hade lätt dysartri och dyspraxi.
Datortomografi och magnetisk resonanstomografi av hjärnan visade normala fynd, liksom rutinblodprov. Likvoranalys visade normalt celltal, normal blod–hjärnbarriärfunktion och inga tecken på intratekal immunglobulinproduktion; koncentrationen av T-tau i likvor var dock mycket kraftigt förhöjd (6810 ng/l), vilket talade för en pågående neuronal destruktion. Likvorkoncentrationen av P-tau var däremot normal (60 ng/l), vilket gav en T-tau/P-tau-kvot på 114, som är betydligt högre än vad som rapporterats vid Alzheimers sjukdom, frontallobsdemens och hos friska kontroller (Figur 1). EEG visade ett icke-konvulsivt status epilepticus med fokus frontotemporalt på höger sida.
Patienten behandlades med höga doser fosfenytoin, sedan i narkos med propofol och senare även med midazolam utan förbättring. EEG visade på generalisering av den epileptiforma aktiviteten, bakgrundsaktiviteten blev alltmer långsam, och trifasiska vågor tillkom. Patienten försämrades hastigt, hon återfick aldrig medvetandet efter sederingen utan avled några månader efter symtomdebut och en månad efter inläggning.
Vid efteranalys av likvor kunde 14-3-3-protein påvisas. Neuropatologisk undersökning bekräftade diagnosen sporadisk Creutzfeldt–Jakobs sjukdom med spongiforma förändringar genom hela kortex och positiv PrPSc-färgning.
Fall 2. En 80-årig, tidigare väsentligen frisk kvinna sökte akut på grund av förvirring, yrsel och gångstörning, som utvecklats under endast några veckor.
I ankomststatus noterades avvikande gångstil, som påminde om skridskoåkning, samt viss »sprittighet«, men inga säkra myoklonier och inget säkert fokalt. Datortomografi med kontrast av hjärnan visade förändringar av den vita substansen. Magnetisk resonanstomografi utfördes inte. Två EEG-undersökningar, den sista 6 månader efter debutsymtom, visade generell förlångsamning men inget för Creutzfeldt–Jakobs sjukdom typiskt mönster. Likvoranalys visade normalt celltal, normal blod–hjärnbarriärfunktion och inga tecken på intratekal immunglobulinproduktion. Analys av 14-3-3-protein var svårbedömd, varför prov skickades till referenslaboratoriet i Edinburgh med negativt resultat. Koncentration av T-tau i likvor var dock mycket kraftigt förhöjd (3598 ng/l), vilket talade för en pågående neuronal destruktion. Koncentrationen av P-tau var normal (70 ng/l), vilket gav en T-tau/P-tau-kvot på 51.
Patienten avled 6 månader efter symtomdebut. Obduktion utfördes, och den neuropatologiska bilden var typisk för Creutzfeldt–Jakobs sjukdom med prionpositiva förändringar. Därutöver påvisades en lindrig alzheimerencefalopati, vilket möjligen förklarar den något lägre T-tau/P-tau-kvoten jämfört med vad som ses i övriga neuropatologiskt verifierade »rena« fall av Creutzfeldt–Jakobs sjukdom (Tabell I). Kvoten är dock fortfarande mycket högre än vad som ses vid enbart Alzheimers sjukdom, frontallobsdemens eller hos friska kontroller (Figur 1).
Fall 3. En 64-årig man som förutom kroniskt förmaksflimmer varit frisk sökte optiker på grund av nedsatt syn. Nya glas hjälpte inte, och symtomen förvärrades. Patienten sökte akut någon månad senare då han plötsligt blivit yr, fått ytterligare försämrad syn och talat osammanhängande.
Vid undersökning på akutmottagningen noterades felpekning med vänster hand vid finger–nästest, men för övrigt var status normalt. Datortomografi av hjärnan var utan anmärkning. Även minnesfunktionen var normal. Symtomen avklingade, och patienten åkte hem påföljande dag. Han återkom 3 veckor senare på grund av försämrat allmäntillstånd; han var då desorienterad, hade intentionstremor och ataktisk gång.
Datortomografi och magnetisk resonanstomografi av hjärnan var utan anmärkning. EEG visade generell förlångsamning och inslag av bi- och trifasiska vågor med frontal dominans, vilket förde tankarna till Creutzfeldt–Jakobs sjukdom. Likvoranalys visade normalt celltal, normal blod–hjärnbarriärfunktion och inga tecken på intratekal immunglobulinproduktion. Däremot påvisades förekomst av 14-3-3-protein. Koncentration av T-tau i likvor var mycket kraftigt förhöjd (4000 ng/l), medan koncentrationen av P-tau var normal (25 ng/l). Detta gav en kraftigt förhöjd T-tau/P-tau-kvot på 160.
Patienten försämrades hastigt, fick kramper och avled 4 månader efter symtomdebut. Obduktion verifierade diagnosen sporadisk Creutzfeldt–Jakobs sjukdom.
Fall 4. En 65-årig, nyligen pensionerad man insjuknade med balanssvårigheter, som av distriktsläkare tolkats som ortostatism. Datortomografi med kontrast av hjärnan 2 månader senare visade normala fynd, men vid återbesök hade symtomen progredierat med gångsvårigheter och minnesstörning. Patienten remitterades till neurologisk klinik med frågeställning Parkinsons sjukdom.
Vid inläggningen noterades ataxi, men inga pareser. Patienten var personlighetsförändrad, orienterad till person och rum, men inte till tid. Magnetisk resonanstomografi av hjärnan visade inga patologiska förändringar. EEG var väsentligen normalt utan fokalitet eller periodiska skarpa vågkomplex. Eftersom patienten försämrades kraftigt utreddes vidare med Creutzfeldt–Jakobs sjukdom som differentialdiagnos.
Vid initial likvoranalys, som utföll med normala fynd vad gäller celltal och blod–hjärnbarriärfunktion, påvisades 14-3-3-protein samt kraftigt förhöjd koncentration av T-tau (1530 ng/l). Koncentration av P-tau analyserades inte vid detta provtagningstillfälle. Vid förnyad lumbalpunktion 3 månader senare hade koncentrationen av T-tau stigit till 3880 ng/l. P-tau var normalt (31 ng/l), vilket gav en kraftigt förhöjd T-tau/P-tau-kvot på 125.
Patienten avled 10 månader efter symtomdebut. Misstanken om Creutzfeldt–Jakobs sjukdom verifierades genom obduktion.
Kommentar
I Tabell I redovisas resultaten från ytterligare 4 neuropatologiskt verifierade fall av Creutzfeldt–Jakobs sjukdom (fall 5–8), varav vi i ett fall inte funnit förekomst av 14-3-3-protein vid den initiala analysen. Samtliga uppvisade en hög T-tau/P-tau-kvot. De beskrivna fallen visar den heterogena sjukdomsbilden, särskilt tidigt i sjukdomens förlopp. Även om diagnosen är ovanlig, ingår den ofta i det differentialdiagnostiska tänkandet, och den bör övervägas vid varje form av hastigt progredierande neurologisk eller psykisk sjukdom. Likvoranalys av 14-3-3-protein är väsentlig, eftersom ett positivt fynd flyttar patienten från möjligt till sannolikt fall enligt WHO-kriterierna (Fakta).
Emellertid är närvaro av 14-3-3 inte specifikt för Creutzfeldt–Jakobs sjukdom [13-16], och sensitiviteten har nyligen ifrågasatts i en studie där endast drygt hälften av patienter med neuropatologiskt verifierad Creutzfeldt–Jakobs sjukdom hade positiv reaktion för 14-3-3 [27]. Metodiken har dock nyligen förfinats, och den tidigare polyklonala antikroppen mot 14-3-3 är nu utbytt mot en monoklonal antikropp, som uppvisar högre affinitet och specificitet. Detta stöds av att proteinet påvisades i fall 2 och 5 vid icke-blind efteranalys med denna metodik.
De ovan redovisade fallen är samstämmiga med nyligen publicerade data, som talar för att bestämning av T-tau/P-tau-kvot i likvor, som komplement till analys av 14-3-3-protein, kan öka möjligheten till säkrare diagnostik vid Creutzfeldt–Jakobs sjukdom [22]. Exakt gränsvärde för T-tau/P-tau-kvoten är svårt att fastställa. I Riemenschneiders och medarbetares studie hade samtliga 20 patienter med Creutzfeldt–Jakobs sjukdom en kvot över 78, och det högsta värdet vid andra demenssjukdomar var 12,8 [22].
Våra 8 fall av Creutzfeldt–Jakobs sjukdom hade alla en kvot över 50 (Tabell I). I en stor studie inkluderande 161 patienter med Alzheimers sjukdom och andra demenssjukdomar fann vi dock 4 fall (Alzheimers sjukdom, frontotemporal demens och Lewy body-demens) med T-tau/P-tau-kvot över 12,8 (13,9–20,8) [28]. Opublicerade data från en studie inkluderande över 250 fall som analyserats vid laboratoriet för klinisk kemi, sektionen för neurokemi, Sahlgrenska Universitetssjukhuset/Mölndal, visar också att enstaka fall av olika demenssjukdomar (Alzheimers sjukdom och Lewy body-demens) liksom andra neurologiska sjukdomar (multipel systematrofi) kan ha en T-tau/P-tau-kvot mellan 13 och 23. Vi anser därför att ett rimligt gränsvärde för T-tau/P-tau-kvoten kan vara 25.
En nyligen uppmärksammad svårighet med tolkningen av biokemiska analysresultat vid misstänkt Creutzfeldt–Jakobs sjukdom är att både 14-3-3-positivitet och T-tau-nivåer beror på sjukdomsdurationen [11]. De högsta T-tau-nivåerna och starkaste 14-3-3-signalerna sågs i denna studie cirka 3–6 månader efter symtomdebut, medan lägre nivåer sågs både sent och tidigt i sjukdomsförloppet [11]. Upprepade lumbalpunktioner kan därför vara av värde om den kliniska bilden inger fortsatt misstanke om Creutzfeldt–Jakobs sjukdom.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
*
ST-läkare Erika Mårtensson, överläkare Gunnar Hallgren, överläkare Nils-Olof Hagnelius, överläkare Harald Karlsson och överläkare Hans Thostrup har bidragit med datainsamling.
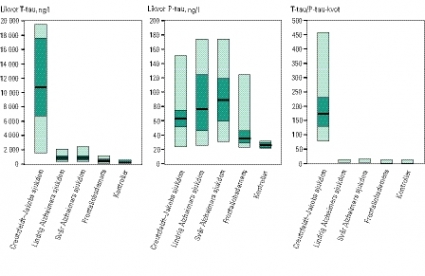
Figur 1. Resultat av likvoranalyser av T-tau, P-tau och T-tau/P-tau-kvoter i likvor från 20 patienter med Creutzfeldt–Jakobs sjukdom, 29 patienter med lindrig Alzheimers sjukdom, 42 patienter med måttlig till svår Alzheimers sjukdom, 18 patienter med frontallobsdemens och 43 kontroller utan CNS-sjukdom [22]. Resultaten är omräknade från pmol/l till ng/l, och vi har inverterat den kvot Riemenschneider och medarbetare [22] rapporterade (P-tau/T-tau). Horisontella linjer indikerar medelvärden, 25:e och 75:e percentilen samt variationsvidd.
Referenser
1. Prusiner SB. Shattuck lecture – neurodegenerative diseases and prions. N Engl J Med 2001;344:1516-26.
3. Lundberg PO. Creutzfeldt-Jakob disease in Sweden. J Neurol Neurosurg Psychiatry 1998;65:836-41.
8. Blennow K, Lind B, Andersson E, Andreasen N. Likvoranalyser i klinisk diagnostik av Creutzfeldt–Jakobs sjukdom. En litteraturgenomgång och tre fall från den kliniska vardagen. Läkartidningen 2001;98:2446-51.
9. Poser S, Mollenhauer B, Kraubeta A, Zerr I, Steinhoff BJ, Schroeter A, et al. How to improve the clinical diagnosis of Creutzfeldt-Jakob disease. Brain 1999;122: 2345-51. 10. Hsich G, Kenney K, Gibbs CJ, Lee KH, Harrington MG. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med 1996;335:924-30.
11. Van Everbroeck B, Quoilin S, Boons J, Martin JJ, Cras P. A prospective study of CSF markers in 250 patients with possible Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2003;74:1210-4.
12. Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000;55:811-5.
13. Burkhard PR, Sanchez JC, Landis T, Hochstrasser DF. CSF detection of the 14-3-3 protein in unselected patients with dementia. Neurology 2001;56:1528-33.
14. Chapman T, McKeel DW Jr, Morris JC. Misleading results with the 14-3-3 assay for the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000;55:1396-7.
15. Saiz A, Graus F, Dalmau J, Pifarre A, Marin C, Tolosa E. Detection of 14-3-3 brain protein in the cerebrospinal fluid of patients with paraneoplastic neurological disorders. Ann Neurol 1999;46:774-7.
17. Otto M, Wiltfang J, Tumani H, Zerr I, Lantsch M, Kornhuber J, et al. Elevated levels of tau-protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Neurosci Lett 1997;225:210-2.
18. Blennow K, Wallin A, Ågren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol 1995;26:231-45.
19. Otto M, Wiltfang J, Cepek L, Neumann M, Mollenhauer B, Steinacker P, et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2002;58:192-7.
20. Van Everbroeck B, Green AJ, Vanmechelen E, Vanderstichele H, Pals P, Sanchez-Valle R, et al. Phosphorylated tau in cerebrospinal fluid as a marker for Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2002;73:79-81.
21. Blennow K, Minthon L, Wallin A. Nya rön om Alzheimers sjukdom: Gott hopp om att biokemiska markörer kan skärpa diagnostiken. Läkartidningen 2000;97:6-10.
22. Riemenschneider M, Wagenpfeil S, Vanderstichele H, Otto M, Wiltfang J, Kretzschmar H, et al. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt-Jakob disease from other dementias. Mol Psychiatry 2003; 8:343-7.
23. Blennow K, Hampel H. CSF markers for incipient Alzheimer´s disease. Lancet Neurol 2003;2:605-13.
24. Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjögren M, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett 2000;285:49-52.
27. Geschwind MD, Martindale J, Miller D, DeArmond SJ, Uyehara-Lock J, Gaskin D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol 2003;60:813-6.
28. Hampel H, Buerger K, Zinkowski R, Teipel SJ, Goernitz A, Andreasen N, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry 2004;61:95-102.
Summary
SUMMARY
Creutzfeldt–Jakob disease (CJD) is a prion disease characterized by rapid neurodegeneration that leads to the death of the patient within months to a few years. Since the disease is transmissible, there is an obligation in Sweden to report possible CJD cases to the Swedish Institute for Infectious Disease Control. To make a diagnosis of CJD is difficult, especially early in the course of the disease when the clinical features may be very vague and heterogeneous. Hence, accurate biological markers both for confirming and excluding CJD would be of great value. The currently recommended investigation of a patient with possible CJD comprises clinical evaluation, electroencephalography, computed tomography or magnetic resonance imaging of the brain and test for 14-3-3 protein in the cerebrospinal fluid (CSF). Recent studies suggest that analysis of total tau (T-tau) and phospho-tau (P-tau) in CSF is a valuable complement to this set of investigations. Here, we review how CSF T-tau and P-tau may aid in the diagnosis of CJD and illustrate this by presenting cases from routine clinical practice.
Henrik Zetterberg, Anna-Lena Hammarin, Petra Nilsson, Elsa Andersson, Börje Lind, Kaj Blennow
Correspondence: Henrik Zetterberg, Laboratoriet för klinisk kemi, Sahlgrenska Universitetssjukhuset, SE-413 45 Göteborg, Sweden
(henrik.zetterberg@clinchem.gu.se)