Sammanfattat
Idiopatiska kardiomyopatier har tack vare det senaste decenniets genetiska landvinningar till stor del kunnat förklaras, och fler än 20 olika gener har visat sig kunna ge upphov till hereditär kardiomyopati.
Ett visst mönster kan urskiljas i vilka gener som ger upphov till hypertrof respektive dilaterad kardiomyopati. Vissa gener kan orsaka båda formerna, men detta kan delvis förklaras av vilken del av genen som muterats.
En stor utmaning blir nu att finna det inbördes förhållandet mellan gendefekt och klinisk diagnostik och behandling. I Sverige har därför ett nationellt register upprättats för att samla in kliniska data och blodprov från barn med primär kardiomyopati.
Hjärtsvikt som följd av primär hjärtmuskelsjukdom, kardiomyopati, är en allvarlig sjukdom och utgör en viktig orsak till plötslig hjärtdöd hos unga och till allvarligt nedsatt funktionsförmåga hos unga och äldre.
För tio år sedan var kardiomyopati en sjukdom som oftast beskrevs som idiopatisk, och spekulationer om orsaken rörde sig ofta kring toxiska, metabola och infektiösa faktorer. Under 1990-talet kom – med förbättrade genetiska och molekylärbiologiska hjälpmedel samt ett tilltagande sökande bland mer eller mindre asymtomatiska familjemedlemmar till drabbade patienter – en ökad kunskap om det betydande inslaget av ärftlighet. Antalet funna genmutationer har nu blivit så betydande att man kan börja urskilja mönster för vilka gener som orsakar olika kliniska former av kardiomyopati. En sammanfattning av dessa forskningsresultat är därför på sin plats.
Klassifikation av kardiomyopatier
Kardiomyopati definieras av WHO/ISFC 1995 [1] som nedsatt hjärtfunktion primärt orsakad av sjukdom i myokardiet (hjärtmuskeln). Kardiomyopati indelas i fyra grupper:
Dilaterad kardiomyopati kännetecknas av vidgning av framför allt vänster kammare, som också uppvisar försämrad kontraktionskraft. Kan vara idiopatisk eller orsakas av genetiska, virala eller toxiska faktorer.
Hypertrof kardiomyopati kännetecknas av vänster- och/eller högerkammarhypertrofi, den är vanligen asymmetrisk och engagerar ibland endast septum. Ofta innebär muskelhypertrofin en förträngning av vänster kammares utflödesområde, varför en tryckgradient uppstår till aorta, sk hypertrof obstruktiv kardiomyopati. Hereditära former överväger.
Restriktiv kardiomyopati kännetecknas av minskad fyllnadsvolym i kamrarna, trots i stort sett intakt systolisk funktion och normal kammarväggstjocklek. Interstitiell fibros är vanlig. Oftast idiopatisk, men finns också associerad med exempelvis amyloidos.
Arytmogen högerkammarkardiomyopati karakteriseras av progressiv fett- och fibrosomvandling av hjärtmuskelcellerna i höger kammare. Senare kan även vänster kammare engageras, men septum sparas ofta. Familjära former är vanliga i denna grupp.
Förutom dessa definierade kardiomyopatier finns ett flertal sjukdomar i hjärtat som inte kan inordnas i dessa fyra grupper, sk oklassificerade kardiomyopatier. Kardiomyopatier finns också associerade med kända generella sjukdomar och kallas då specifika kardiomyopatier. I denna grupp ingår olika kardiovaskulära, metabola, endokrina och inflammatoriska tillstånd.
I denna översiktsartikel betonas de primära kardiomyopatier som på senare år fått genetisk och molekylärbiologisk förklaring. Dessa finner man huvudsakligen i grupperna dilaterad respektive hypertrof kardiomyopati. Vi har dessutom fokuserat på de gener som kodar för strukturella proteiner, eftersom mutationer i dessa utgör den största delen av ärftliga former av kardiomyopati.
Epidemiologi
Hypertrof kardiomyopati är den vanligaste formen av kardiomyopati. I en amerikansk undersökning har visats en prevalens på 200/100000 individer [2], medan dilaterad kardiomyopati har en prevalens på 36,5/100000 [3]. I barnpopulationen har en amerikansk undersökning, baserad på ett register över nya fall av kardiomyopati bland barn i åldern 1–18 år i sydvästra och nordöstra USA (1996–1999), visat en årlig incidens på 1,3/100000: varav dilaterad kardiomyopati 51procent, hypertrof kardiomyopati 42 procent och övriga 7 procent [4]. Delas denna grupp upp i barn 0–1 år och barn 1–18 år blir incidensen 8,34 respektive 0,70/100000, vilket visar att första levnadsåret helt överväger för insjuknande i kardiomyopati. En finsk retrospektiv nationell studie av barn i åldern 0–20 år som insjuknat 1980–1991 visar liknande incidens och prevalens [5].
I en svensk studie av yngre vuxna, 15–35 år, som dött en plötslig hjärtdöd utan föregående känd sjukdom, har man visat att orsaken varit dilaterad kardiomyopati i 12,2 procent och hypertrof kardiomyopati i 10,5 procent av fallen [6]. Om vi kan hitta tidiga markörer för dessa sjukdomar, kliniska eller genetiska, framför allt hos dem där sjukdomen är känd i familjen, skulle ett antal av dessa dödsfall rimligen kunna förhindras genom tidig farmakologisk behandling eller insättning av intrakardiell defibrillator. Wisten och medarbetare har följt upp denna studie med att undersöka EKG-förändringar (hos de 66 av 162 där EKG kunde återfinnas) och fann patologiska förändringar i 82 procent av fallen [7]. Vi kommer här att visa att genetiska markörer kan komma att spela en viktig roll för att finna möjliga högriskpatienter.
Hjärtmuskelns funktion
För att förstå de genetiska landvinningar som gjorts inom kardiomyopatiforskningen följer nedan en översiktlig beskrivning av hur hjärtmuskelcellen fungerar på cellulär nivå. För närmare beskrivning av de proteiner som berörs, se Fakta.
Sarkomeren – hjärtats motor.
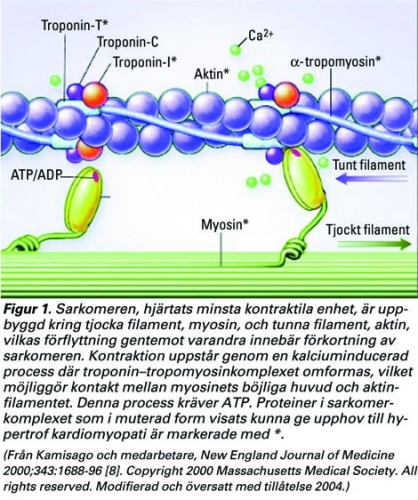
Hjärtats funktion, att pumpa blod till lungorna och kroppen, förutsätter en förmåga att cykliskt förändra höger och vänster kammares volym. Den minsta molekylära enhet som gör detta möjligt, hjärtats motor, är sarkomeren (Figur 1). Denna enhet genererar kraften som leder till hjärtmuskelfibrernas förkortning, kontraktion.
Sarkomerens kontraktila element är uppbyggt av tjocka filament, bestående av myosin, och tunna filament, bestående av aktin. Andra viktiga proteiner som associerar till dessa i sarkomeren är troponinkomplexet och a-tropomyosin. Vid kontraktion klättrar myosin längs aktin, och på detta sätt förkortas hjärtmuskelfibrerna (Figur 1). Denna process är beroende av energi (ATP) och kalcium. Titin, kroppens största protein, sträcker sig längs hela sarkomeren och utgör en viktig del i kontraktionen, en slags fjäder som står för sarkomerens elasticitet och dessutom deltar i kraftöverföringen till cellskelettet.
En förutsättning för att hjärtmuskelcellen verkligen förkortas av förändringen i sarkomerens längd är att kraften överförs till cellmembranet och ut till extracellulära vävnaden. Detta sker genom en kedja av proteiner, där varje protein utgör en nödvändig länk för att optimal kraftöverföring skall ske (Figur 2). Desmin och dystrofin är exempel på sådana proteiner som förankrar sarkomeren till cellmembranet och till extracellulära proteinmatrix via länkande proteiner. Strukturella fel i någon av dessa länkar ger upphov till försämrad hjärtmuskelfunktion, kardiomyopati.
Kalcium – tändgnistan.
Kalcium är den trigger som kan utlösa en kontraktion i hjärtmuskelcellen. Det finns därför ett noggrant reglerat system, som styr kalciumflödena i cellen. I cellmembranet finns kalciumkanaler av L-typ (benämnda DHPR [dihydropteridinreduktas]), och som svar på en aktionspotential, ursprungligen utgången från sinusknutan, släpper de in en liten mängd kalcium i cellen (Figur 2). Denna relativt lilla koncentrationshöjning av kalcium aktiverar kalciumkanaler i sarkoplasmatiska retiklet (sk ryanodinreceptorer, RyR) som släpper ut en stor mängd kalcium från detta kalciumlager till cytoplasman där det aktiverar kontraktionen genom att binda till troponin-C. När kalcium binds till troponin-C förs hela troponin–tropomyosinkomplexet åt sidan på tunna filamentet och möjliggör bindning av myosin, som med ATP som energi således kan klättra fram på aktinfilamentet (kontraktion, Figur 1).
När sedan hjärtmuskeln skall relaxera under diastole måste kalciumkoncentrationen åter minska, och då stängs ryanodinreceptorerna; i stället öppnas andra kalciumkanaler, sk SERCA (sarcoplasmic/endoplasmic reticulum calcium ATPase), som under inflytande av proteinet fosfolamban återför kalcium till sarkoplasmatiska rummet. Mutationer i de gener som kodar för ryanodinreceptorn och fosfolamban har visats kunna ge upphov till kardiomyopati.
Hypertrof kardiomyopati
Klinik och patologi. Den kliniska bilden varierar från i princip symtomfrihet till palpitationer, hjärtrytmrubbningar, bröstsmärta, synkope och kan sedan leda till hjärtsvikt och plötslig död. Hjärtats kontraktilitet är oftast initialt normal eller till och med ökad, men relaxationsfasen (diastole) är försämrad på grund av stelt myokardium. Detta leder till förmaksdilatation och vänstersidig hjärtinkompensation.
Diagnosen baseras huvudsakligen på ekokardiografi, som visar hypertrofi av vänster kammare, antingen symmetrisk, dvs omfattande hela hjärtat, eller asymmetrisk omfattande endast kammarskiljeväggen. Histologiskt ses förstorade hjärtmuskelceller, myofibrillär disorganisation och bindvävsomvandling. Den senare bidrar till hjärtats oförmåga att relaxera på ett normalt sätt i diastole.
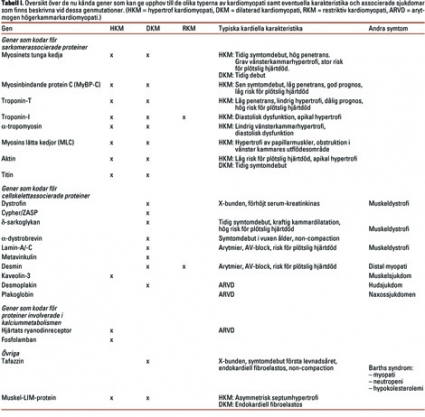
Genetik. Hypertrof kardiomyopati är den vanligaste ärftliga kardiovaskulära sjukdomen, och två tredjedelar av fallen bedöms ha en autosomalt dominant ärftlighet. Man har funnit 12 gener i vilka mutationer ger upphov till hypertrof kardiomyopati, och 10 av dessa kodar för proteiner som är direkt involverade i hjärtats kontraktila enhet, sarkomeren. Således tycks hypertrof kardiomyopati huvudsakligen vara en konsekvens av dysfunktion i hjärtmuskelfibrernas kontraktionsförmåga, kraftgenerering.
Djurförsök har påvisat att man kan se tecken på nedsatt hjärtfunktion före utvecklingen av hypertrofi, varför man spekulerat över om hypertrofin i hjärtmuskeln är en sekundär effekt. Hypertrofi kan ju även ses vid extrem belastning på hjärtat vid exempelvis hypertoni, och samma funktionella processer skulle kunna kopplas in oavsett om det rör sig om en primär eller en sekundär hjärtsjukdom. Mekanismen för detta kan vara att den nedsatta kontraktiliteten i hjärtmuskelcellerna leder till ett stressvar och en aktivering av tillväxtfaktorer, som sedan bidrar till hypertrofi av hjärtmuskelcellerna och tillväxt av bindvävsceller – fibros.
Man har tidigare bedömt risken för plötslig hjärtdöd hos patienter med hypertrof kardiomyopati dels på anamnestiska uppgifter om tidig debut, svimningstillbud och plötslig död i familjen, dels på förekomst av patologisk blodtrycksreaktion vid arbetsprov eller maligna arytmier. Kanske kan specifik information om vilken gen och vilken mutation som är involverade bli ytterligare ett redskap i denna riskbedömning?
Mutationer vid hypertrof kardiomyopati som är associerad med plötslig hjärtdöd kännetecknas oftast kliniskt av att de resulterar i kraftig kammarhypertrofi, hög penetrans och tidig debut. Ingen klinisk bild är typisk för en viss mutation, och en specifik genmutation kan ge upphov till olika kliniska symtom. Myosin är i många studier det protein som oftast är muterat, i cirka en tredjedel av fallen. Myosinbindande protein C (MyBP-C), ett reglerande protein i sarkomeren, är näst vanligast och återfinns muterat i 20–30 procent av fallen, följt av troponin-T med 5–10 procent. Övriga gener svarar för 5 procent av fallen. Generellt kan sägas att hypertrof kardiomyopati som orsakas av mutationer i myosin debuterar i yngre ålder, ger mer extensiv hypertrofi och högre incidens av plötslig hjärtdöd än hypertrof kardiomyopati som orsakas av mutationer i MyBP-C. Troponin-T-mutationer ger lindrig hypertrofi och har låg penetrans, men trots detta har patienter med dessa mutationer klart ökad mortalitet. Således skulle det vara av speciellt värde att utesluta eller bekräfta mutationer i denna gen, för både patienten och dennes släktingar, eftersom man här inte kan gå efter kliniska fynd i riskbedömningen.
I en undersökning från Umeå screenades 46 patienter från norra Sverige med familjära och sporadiska former av hypertrof kardiomyopati för mutationer i de 8 vanligaste gener som orsakar denna form av kardiomyopati. Samtliga dessa gener kodar för sarkomerproteiner [10]. Man fann 11 olika mutationer, varav 6 aldrig tidigare beskrivna, hos 13 patienter, dvs nästan en tredjedel. Ett intressant fynd i denna studie var att den vanligaste muterade genen var MyBP-C, inte myosin som tidigare beskrivits från andra populationer. Detta har betydelse, eftersom kardiomyopati orsakad av en MyBP-C-mutation har senare debut och lägre penetrans än den orsakad av myosinmutationer, vilket medför ökad risk i MyBP-C-gruppen för förekomst av dold sjukdom i släkten. Släktingar till en patient med denna mutation bör troligen gå på kontroller långt upp i medelåldern, eftersom hypertrofin ibland ger sig till känna först vid 40–50 års ålder.
I ett nyligen utkommet konsensusdokument utformat av en expertpanel bestående av kardiologer från Europa och USA finns en mer detaljerad genomgång av diagnostik, behandling och prognos för patienter med hypertrof kardiomyopati [11]. En behandlingsform som visats effektiv är implantation av intrakardiell defibrillator. I en multicenterstudie av patienter med hypertrof kardiomyopati och hög risk för plötslig hjärtdöd och som behandlats med intrakardiell defibrillator kunde man visa att under en uppföljningstid på i genomsnitt 3 år förhindrades potentiellt livshotande arytmier i 25 procent av fallen [12]. I de fall där intrakardiell defibrillator inte är tillämplig eller hos barn som är för unga för denna behandlingsmetod kan lågdos amiodaron ha skyddande effekt.
Dilaterad kardiomyopati
Klinik och patologi. Dilaterad kardiomyopati är ett allvarligt sjukdomstillstånd med hög mortalitet och den vanligaste orsaken till hjärttransplantation, 5-årsmortaliteten är cirka 50 procent [13]. Symtomen är initialt ofta ospecifika med trötthet, dyspné och hjärtklappning, men med försämrad kontraktil funktion följer hjärtsvikt och hjärtrytmrubbningar. Vid dilaterad kardiomyopati är den histologiska bilden mer ospecifik än vid hypertrof kardiomyopati, och den kan vara helt normal. Man kan dock i vissa fall se tecken på hypertrofi eller degeneration av hjärtmuskelceller samt bindvävsomvandling. Diagnosen ställs oftast med ekokardiografisk undersökning, där man ser förstorad kammarvolym och ofta nedsatt kontraktionsförmåga. En sådan ekokardiografisk bild kan ha många bakomliggande orsaker, och man måste utesluta bla koronarkärlssjukdom, myokardit, sköldkörtelrubbning och toxisk påverkan innan man kan ställa diagnosen primär distal kardiomyopati och börja leta efter genetiska orsaker. Anamnes på andra fall i familjen har naturligtvis stor betydelse.
Genetik. I en studie av Michels och medarbetare på patienter med dilaterad kardiomyopati befanns >20 procent ha en förstagradssläkting med sjukdomen [3]; i en australisk studie på barn hade 15 procent distal kardiomyopati i familjen, och dessa fall hade också signifikant tidigare symtomdebut [14]. Såväl autosomalt dominant, autosomalt recessivt, X-bundet och mitokondriellt ärftlighetsmönster har beskrivits.
Till skillnad från hypertrof kardiomyopati, där den genetiska defekten oftast härrör från gener som kodar för proteiner involverade i sarkomerens funktion, har distal kardiomyopati en mer heterogen genetisk bakgrund. När de första mutationerna som orsakar dilaterad kardiomyopati upptäcktes i mitten av 1990-talet verkade de flesta muterade gener koda för proteiner som på något sätt överför kraften från sarkomeren till hjärtmuskelcellen. Man trodde då att man funnit en gemensam faktor för mekanismen bakom hjärtdilatationen. När fler kardiomyopatiassocierade gener beskrevs blev dock bilden mer komplicerad: man upptäckte att mutationer av gener som tidigare beskrivits vara förändrade vid hypertrof kardiomyopati även kunde leda till distal kardiomyopati (Tabell I).
I den övervägande andelen av hereditära dilaterade kardiomyopatier kan man dock finna orsaken i muterade gener som kodar för proteiner som förankrar sarkomeren i hjärtcellen. Således ger en försämrad kraftöverföring upphov till att kammaren dilateras och inte till någon kompensatorisk hypertrofi. I Figur 2 visas de viktigaste proteinerna som är involverade i kraftöverföringen från sarkomeren till omgivande vävnad, de gener som ger dilaterad kardiomyopati när de är muterade är markerade med #.
Det finns ytterligare fynd som stödjer hypotesen att sviktande hjärtmuskel beror på defekter i cellskelettet. Coxsackievirus, som man vet kan orsaka dilaterad kardiomyopati, har ett enzym som kan bryta ned dystrofin och som har visat sig göra det i infekterade hjärtmuskelceller [15]. Man har också sett att dystrofin är derangerat hos patienter med grav hjärtsvikt med kammardilatation, oavsett genes. Det intressanta är att man här har kunnat visa att dystrofin normaliseras efter avlastning med mekaniskt hjärta [16].
Andra gener som kan ge upphov till distal kardiomyopati är tafazzin och lamin-A/-C (Fakta), men här är mekanismen mer oklar.
Mutation i samma gen – hypertrof eller distal kardiomyopati
Av de gener som kan ge hypertrof kardiomyopati finns hittills 9 beskrivna som även kan ge distal kardiomyopati. Hur kan det vara möjligt? I vissa fall rör det sig om olika mutationer som ger olika typ av kardiomyopati, och man skulle då kunna förklara detta med att mutationen drabbar olika funktionella delar av proteinet. Detta kan exemplifieras av mutationer i genen aktin där mutationer som involverar regioner där aktin interagerar med myosin, och därmed direkt involverar muskelkontraktionen, ger upphov till hypertrof kardiomyopati, medan mutationer som involverar aktinets bindning till cellskelettproteiner ger upphov till distal kardiomyopati [17]. Likaså har olika mutationer i de reglerande proteinerna i tunna filamentet, troponin och tropomyosin, olika funktionella konsekvenser. De mutationer som ger hypertrof kardiomyopati ger upphov till ökad kalciumkänslighet jämfört med de normala proteinerna, och de mutationer som ger distal kardiomyopati ger minskad kalciumkänslighet; i olika mätningar på muskelcellernas funktion ger de ökad respektive minskad kontraktilitet [18] [Charles Redwood, London, pers medd, 2003].
I andra fall kan man dock inte förklara skillnaderna i klinisk bild på detta sätt, och det finns till och med patienter inom samma familj som har samma gendefekt men olika sjukdomsuttryck. Här får man snarare tänka sig att miljö (tex träningsgrad), varierande fysiologiska faktorer (tex hypertoni) eller genetisk bakgrund (modifierande gener) kan ha betydelse för om mutationen resulterar i distal eller hypertrof kardiomyopati.
Restriktiv kardiomyopati
Restriktiv kardiomyopati utgör en klar minoritet i gruppen kardiomyopatier och kännetecknas av försämrad kammarfyllnad och nedsatt diastolisk funktion utan tecken på dilatation eller hypertrofi av kamrarna. Histopatologiskt ses ofta fibrosomvandling i hjärtmuskeln. De flesta fall av restriktiv kardiomyopati är sporadiska, men familjära fall finns beskrivna, och då även i familjer där majoriteten av de sjuka har hypertrof kardiomyopati. Således kan man ifrågasätta om restriktiv kardiomyopati är en egen genetisk entitet, och man har ännu inte funnit någon mutation som ger enbart restriktiv kardiomyopati. De gener som hittills visats vara muterade vid restriktiv kardiomyopati är desmin [19] och troponin-I [20].
Arytmogen högerkammarkardiomyopati
Arytmogen högerkammarkardiomyopati (arrhythmogenic right ventricle dysplasia) är en diagnos som är svår att ställa kliniskt, och den debuterar ofta i form av ventrikulär eller supraventrikulär arytmi. Patologiskt kännetecknas arytmogen högerkammarkardiomyopati av celldöd i hjärtmuskeln och infiltration av fettceller i framför allt höger kammares muskelvägg, vilket ofta ger ett karakteristiskt utseende vid magnettomografiundersökning av hjärtat. De ärftliga formerna, cirka 30 procent av fallen, är oftast autosomalt dominanta, men även autosomalt recessiva fall finns beskrivna. Mutationer i genen kodande för ryanodinreceptorn i hjärtmuskeln ger upphov till arytmogen högerkammarkardiomyopati. Ryanodinreceptorn är involverad i hjärtcellens kalciumomsättning (se ovan), och störningar i denna skulle kunna ge upphov till celldöd. Vid den autosomalt recessiva Naxossjukdomen, som karakteriseras av arytmogen högerkammarkardiomyopati, hårförändringar och palmoplantar keratos, har man funnit mutationer i de gener som kodar för desmoglobin och plakoglobin, vilka båda har som funktion att sammanbinda cellskelettet med cellmembranet i de sk desmosomerna. En defekt i desmosomerna kan ge skador i celler som utsätts för mekanisk stress, vilket skulle förklara den celldöd som sker i hjärtcellerna.
Kardiomyopatier och skelettmuskelsjukdomar
Eftersom både hjärtmuskelns och skelettmuskelns celler är tvärstrimmiga, dvs har samma uppbyggnad som sarkomerer som förankras via cellskelettet, är många proteiner i dessa vävnader gemensamma.
Det innebär att patienter med ärftliga skelettmuskelsjukdomar, tex muskeldystrofier och primära myopatier med mutationer i sådana strukturella muskelproteiner (tex dystrofin, sarkoglykan eller desmin), också ofta har hjärtmuskelengagemang. Detta är viktigt att känna till – i synnerhet som patienter med muskelsjukdom på grund av muskelsvaghet ofta är så litet fysiskt aktiva att hjärtsvikt kan utvecklas »tyst« och ge sig till känna först i ett sent skede, samtidigt som tidigt insatt hjärtsviktsbehandling anses kunna påverka förlopp och prognos. I det stora flertalet fall rör det sig här om dilaterad kardiomyopati.
Omvänt är det känt att patienter med hjärtsvikt kan vara muskelsvaga, och mycket talar för att det finns sekundära störningar i deras metabolism som ger upphov till denna svaghet. Det är dock viktigt att ha dold muskelsjukdom i åtanke, liksom att det kan finnas en gemensam genetisk bakgrund till både hjärt- och muskelsymtomen.
Relation mellan symtombild och gendefekt
Har de framsteg som gjorts inom den molekylära kardiologin någon klinisk relevans? För att kunna besvara denna fråga måste vi börja koppla samman kliniska undersökningsfynd med de mutationer som beskrivits översiktligt här. Man har redan kunnat visa att mutationer i vissa gener ger mer allvarlig form av kardiomyopati än mutationer i andra. Exempelvis orsakar mutationer i myosin ofta en måttlig till svår form av hypertrof kardiomyopati med hög genetisk penetrans, medan mutationer i troponin-T ofta orsakar en lindrig form av hypertrof kardiomyopati eller inga kliniska symtom alls. Vissa mutationer har också i större utsträckning visat sig vara associerade med plötslig hjärtdöd, och man kan här även se en skillnad mellan olika mutationer inom samma gen. Detta kan ge vägledning för prognosen samt hur aktiv man skall vara med uppföljning och eventuell behandling, såsom medicinering eller intrakardiell defibrillator.
Denna relativt unga kardiologiska genetik kommer säkerligen framöver att utnyttjas kliniskt i allt större utsträckning, på samma vis som skett när det gäller genetiska orsaker till neuromuskulära sjukdomar.
På initiativ av docent Daniel Holmgren, Drottning Silvias barn- och ungdomssjukhus i Göteborg, sammanställs nu ett nationellt register över barn och ungdomar med kardiomyopati. Där samlas såväl anamnestiska och kliniska data som blodprov för möjlighet till biokemiska och genetiska analyser. Tanken är att kunna knyta samman specifika kliniska karakteristika med biokemiska eller genetiska markörer för att så småningom kunna hitta bättre sätt att diagnostisera kardiomyopati och förhoppningsvis även finna specifika behandlingsmetoder.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.

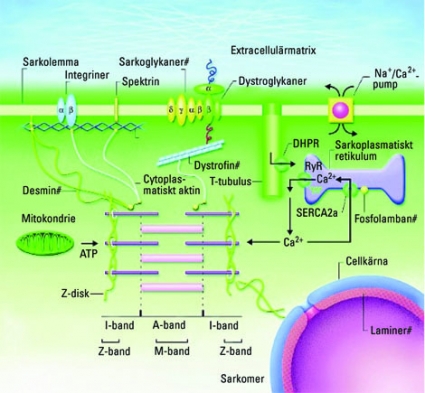
Figur 2. Schematisk skiss av hjärtmuskelcellens uppbyggnad. Centralt i figuren ses sarkomeren, vars tunna aktinfilament förankras på var sida i de s k Z-diskarna. Z-diskarna består av ett flertal proteiner, bl a desmin som också sammanbinder Z-diskarna med varandra, cellkärnan och cellmembranet. Även aktin utgör en brygga mellan sarkomeren och cellens utsida via länkande proteiner som dystrofin och dystroglykaner. Kalcium inducerar muskelkontraktionen genom att rörartade kanaler i cellen, T-tubuli, släpper in små mängder kalcium via proteinet dihydropteridinreduktas (DHPR). Den lilla kalciumkoncentrationsökningen utlöser ett större utflöde av kalcium från sarkoplasmatiska retiklet via proteinet ryanodin (RyR), och sarkomeren aktiveras. Kalciumjonerna återupptas sedan snabbt genom att andra kanaler öppnas: SERCA2a i sarkoplasmatiska retiklet och Na–Ca-pumpen i cellmembranet. Proteiner som i muterad form visat sig kunna ge upphov till dilaterad kardiomyopati är märkta med #.
Referenser
1. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, OConnell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996;93(5):841-2.
2. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995;92(4):785-9.
3. Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992;326(2):77-82.
4. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003;348(17):1647-55.
5. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, et al. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol 1997;146(5):385-93.
6. Wisten A, Forsberg H, Krantz P, Messner T. Sudden cardiac death in 15–35-year olds in Sweden during 1992–99. J Intern Med 2002;252(6):529-36.
7. Wisten A, Andersson S, Forsberg H, Krantz P, Messner T. Sudden cardiac death in the young in Sweden: electrocardiogram in relation to forensic diagnosis. J Intern Med 2004;255(2):213-20.
8. Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 2000;343(23):1688-96.
9. Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev 2002;82(4):945-80. 10. Morner S, Richard P, Kazzam E, Hellman U, Hainque B, Schwartz K, et al. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol 2003;35(7):841-9.
11. Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J 2003;24(21):1965-91.
12. Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med 2000;342(6):365-73.
13. Grogan M, Redfield MM, Bailey KR, Reeder GS, Gersh BJ, Edwards WD, et al. Long-term outcome of patients with biopsy-proved myocarditis: comparison with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 1995;26(1):80-4.
14. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003;348(17):1639-46.
15. Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med 1999;5(3):320-6.
16. Vatta M, Stetson SJ, Jimenez S, Entman ML, Noon GP, Bowles NE, et al. Molecular normalization of dystrophin in the failing left and right ventricle of patients treated with either pulsatile or continuous flow-type ventricular assist devices. J Am Coll Cardiol 2004;43(5):811-7.
17. Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, et al. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest 1999;103(10):R39-43.
18. Robinson P, Mirza M, Knott A, Abdulrazzak H, Willott R, Marston S, et al. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J Biol Chem 2002;277(43): 40710-6.
19. Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 1998;19(4):402-3.
20. Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 2003;111(2):209-16.