Taruis sjukdom beskrivs hos sex personer från två familjer med västerbottniskt ursprung.
Tillståndet är autosomalt, recessivt ärftligt och karakteriseras av ansträngningsintolerans och icke-sfärocytärhemolys.
Fenotypen framkallas av brist på det glykolytiska enzymet fosfofruktokinas, som är av muskeltyp.
Hemolysen kan förklaras av ökad, passiv diffusion av Ca-joner genom erytrocytmembranet med intracellulär Ca++-ökning och minskad celldeformabilitet som följd.
Myopatin kan variera individuellt från helt tolerabel till mycket besvärande.
Med nedanstående fallbeskrivningar vill vi illustrera vikten av att utreda alla icke uppenbara orsaker till ikterus och poängtera att bakom en eventuell Mb Gilbert-diagnos kan dölja sig en annan gendefekt. Även om de terapeutiska möjligheterna är små vid Taruis sjukdom är klarheten om diagnosen många gånger lugnande och förklarande för patienterna, som härigenom får en rationell orsak till att vara trötta och inte kunna orka med det som förväntas av omgivningen.
Risken för felklassificering
Ikterus är ett inte alldeles ovanligt symtom i klinisk praxis. Flera tänkbara orsaker föreligger, och det är viktigt att tidigt utesluta stopp i gallgångar. Till mer ovanliga orsaker hör hemolytisk sjukdom som orsakas av intraerytrocytär enzymbrist i glykolysen och som sällan påträffas i klinisk verksamhet. Den blir ofta felklassificerad eller utan diagnos.
Patienterna är ikteriska, och ett kroniskt hemolytiskt tillstånd framträder under utredningens gång. Eftersom ingen immunhemolys, sfärocytos eller hemoglobinopati kan påvisas blir Mb Gilbert den preliminära diagnosen, som dock hade kunnat undvikas om i detta inledande skede en haptoglobinundersökning också gjorts, eftersom en haptoglobinsänkning oftast utesluter denna diagnos. I detta sammanhang bör emellertid påpekas att det är normalt med icke-mätbara haptoglobinkoncentrationer ända upp i tonåren. Därför är haptoglobinbestämning av ringa intresse vid anemidiagnostik på barn.
Intraerytrocytär enzymopati, Taruis sjukdom
Efter upptäckten av pyruvatkinasbrist i erytrocyterna för mer än 40 år sedan har det visats att bortfall av vart och ett av nästan alla glykolytiska enzymer kan orsaka anemi på basen av ett icke-sfärocytärhemolytiskt tillstånd. Enzymdefekterna är kongenitala och i några former inte begränsade enbart till den erytropoetiska cellinjen utan finns även i ett eller flera organsystem med lokala manifestationer [1, 2]. En sådan »inborn error of metabolism« är brist på fosfofruktokinas (PFK), först beskriven av Tarui och medarbetare [3] som en ansträngningsmyopati med måttlig glykogendeposition i skelettmusklerna (glykogenos VII). De påvisade också en nästan fullständig brist på muskel-PFK, medan en partiell enzymbrist förelåg i erytrocyterna (se nedan). I de röda blodkropparna är PFK-aktiviteten cirka hälften av den normala. Erytrocyterna har förkortad överlevnadstid, och hemolytisk ikterus är det andra framträdande symtomet vid Taruis sjukdom.
Ett 90-tal fall av fosfofruktokinasbrist har rapporterats i litteraturen, och en sammanställning med klassindelning efter fenotyp och debutålder har presenterats [4]. Cirka hälften av de drabbade visar den klassiska fenotypen med tidigt debuterande ikterus och myopati; sen debut finns också beskriven, dock endast från ett fåtal personer. En infantil typ som manifesterar sig vid födelsen och med tidig död har beskrivits. Slutligen har ett mindre antal fall med enbart hemolys publicerats.
Fosfofruktokinasets biokemi
Humant PFK är en tetramer av peptidsubenheter. Tre typer av subenheter finns i kroppens vävnader: muskel- (PFK-M), lever- (PFK-L) och trombocyttyp (PFK-P). Humant muskel- och lever-PFK består av homotetramerer (M4 respektive L4), medan den röda blodkroppens enzym består av fem isoenzymer (M4, M3L, M2L2, ML3 och L4) [5]. Enzymets tre subenheter kodas av gener i skilda kromosomer: M-typen i kromosom 12, L-typen i kromosom 21 och P-typen i kromosom 10 [4, 6].
Nedärvningen vid Taruis sjukdom är autosomalt recessiv, och endast personer med homozygoti eller sammansatt heterozygoti visar sjukdomssymtom. Fosfofruktokinas är ett allostert reglerat enzym, där framför allt ATP, ADP och AMP är effektormolekyler på så sätt att högre nivåer av ATP hämmar enzymaktiviteten, medan högre ADP- och AMP-nivåer stimulerar enzymaktiviteten. Den konventionella synen på Taruis sjukdom har varit att det föreligger ett sänkt glykolytiskt flöde i erytrocyterna på grund av bortfallet av M-subenheten i PFK.
Ny patofysiologisk mekanism visad
Vi har nyligen visat att så inte är fallet. I stället kan – paradoxalt nog – ett ökat glykolytiskt flöde påvisas i erytrocyterna från patienter med Taruis sjukdom [7]. Denna ökade glykolys i erytrocyterna förklaras med ett ökat kalciumläckage in i cellerna vid denna sjukdom [8]. Denna förhöjda kalciumbelastning provocerar ett speciellt Ca++-ATPas att arbeta för högtryck för att pumpa ut Ca++ ur cellerna, och en metabolisk koppling föreligger mellan ATPas-aktiviteten och den glykolytiska aktiviteten. Nettoeffekten blir likväl sänkta nivåer av såväl ATP som 2,3- bisfosfoglycerat (2,3 BPG) medan laktat är stegrat. En sänkning av 2,3-BPG-koncentrationen i erytrocyterna leder till en vänsterförskjutning av syrgasdissociationskurvan, med således minskad syreavgivning till målcellerna i kroppens olika organ och vävnader. Detta leder i sin tur till ökad erytropoietininsöndring med slutresultatet att den hemolytiska anemin oftast blir kompenserad.
Kliniska symtom
Kliniskt framträder myopatin med varierande muskelsvaghet, snabb uttröttbarhet, ibland smärta och kramper samt värk i ländryggen och myoglobinuri. Mattheten kan vara snabbt övergående vid vila men kan kvarstå i dagar efter excessiv ansträngning. Ökad urinsyrabildning med gikt är inte ovanligt [9]. Enstaka fall har upptäckts vid omhändertagande efter rabdomyolys, enbart eller med akut njurinsufficiens som följd [10, 11].
PFK-M-bristens symtom är likartade dem hos patienter med McArdles sjukdom. En väsentlig skillnad är dock att de senare inte hemolyserar. Dessa personer med myofosforylasbrist kan inte spjälka muskelglykogen till glukos men däremot utnyttja blodglukos för ATP- och pyruvat/laktatbildning. Glykolysen ger den energi som behövs vid snabb övergång från vila till arbete samt till pyruvatbildning, som är nödvändig för den vidare oxidativa fosforyleringen av fettsyrornas metaboliter (efter b-oxidationen ) [12].
Fallbeskrivningar
Vi har tidigare beskrivit en släkt i Västerbotten med fosfofruktokinasbrist [13] och kan nu rapportera ytterligare en familj i Jämtland med två afficierade barn.
Västerbottensfamiljen
Probanden (Figur 1 II:3) är näst yngste sonen i en västerbottnisk jordbrukarfamilj med tio nu levande barn. Han har sedan barndomen varit lätt gul i ögonvitorna men inte haft någon läkarkontakt angående detta förrän inskrivningsläkare remitterade honom till sjukvården för utredning vid 18 års ålder. Av sjukhusläkare noterades en opåverkad yngling i gott allmäntillstånd men med gulaktiga sclerae, dock utan tydlig ikterus i huden. Rutinanalyser visade ett högt bilirubinvärde, men övriga leverprov var normala. Retikulocytantalet var ökat, och de röda blodkropparnas osmotiska resistens normal. En sju år äldre bror hade tidigare fått diagnosen Gilberts sjukdom, vilket bidrog till att även probanden fick denna lugnande diagnos. Värnplikten kunde sedan genomföras inom flygvapnets marktjänst utan påtagliga svårigheter.
Ett besvärande och oförklarat handikapp har varit hans muskelsvaghet med tidigt insättande kraftlöshet och kvardröjande trötthet sedan tidiga år, vilket begränsat hans deltagande i lekar och idrotter. Trots stark vilja att förbättra konditionen med joggning och annan muskelträning har det inte gett önskat resultat. Effekten har i stället blivit muskelsmärtor, kramp och ibland värk i ländryggen. Deltagande i någon lagidrott har ibland lett till närmast förlamning av benen, vilken dock släppt efter en stund. Patienten har, trots sitt handikapp, kunnat försörja sig med lättare verkstadsarbete och som taxi- och busschaufför. Vid drygt 20 års ålder tog han ny sjukhuskontakt på grund av gastrit med ont i magen, kräkningar och sura uppstötningar, vilket behandlades med sedvanliga mediciner.
Ungefär tio år senare blev han på nytt inlagd på sjukhus med symtomen ledsmärtor, feber, huvudvärk och snabbt sjunkande hemoglobinvärde. Benmärgsprov visade nästan total frånvaro av erytropoes men i övrigt normal bild. Efter en vecka hade erytrocytbildningen normaliserats; patienten hade troligen drabbats av en parvovirus B 19-infektion.
Vid den fortsatta utredningen vid universitetssjukhuset i Umeå bekräftades bilden av uttalad hemolys men utan klar anemi. Totalbilirubin låg på en hög nivå liksom retikulocyttalet, med motsatt lågt värde för haptoglobin (Tabell I). Erytrocyternas osmotiska resistens (suspension av blodkropparna i olika buffertspädningar) var normal i EDTA-blod. Ingen sfärocytos förelåg, och extrakorpuskulära orsaker till hemolysen kunde uteslutas, varför grundfelet fick sökas under rubriken hereditär icke-sfärocytärhemolytisk anemi i litteraturen.
Probanden är nu i 50-årsåldern med i stort sett oförändrad, uttalad hemolys, hyperbilirubinemi och kraftig retikulocytos. Muskelsvagheten tycks ha förvärrats med tex värk i vadmusklerna vid promenader i uppförsbackar och kvarstående trötthet upp till något dygn efter kraftigare muskelansträngning. Sjukskrivningsperioderna har kommit allt oftare och blivit längre.
Äldre bror. Vid blodprovstagning på syskonen upptäcktes även en äldre bror och en yngre syster (Figur 1, II:2 och II:4) med klara tecken till hemolys. Den äldre brodern hade också haft gulfärgade ögonvitor sedan barndomen, dock utan att detta oroat någon i omgivningen. Han betraktades kroppsligen som klen och spinkig; att mäta sig med bröderna i kroppsarbete var inte möjligt, trots den goda viljan.
Värnpliktsutbildningen genomfördes vid luftvärnet utan särskild anmärkning från befäl. Långsamhet eller trötthetsyttringar vid förflyttningar uppfattades dock som lättja eller maskning. En speciell episod som han särskilt minns inträffade vid löpträning med packning. Efter någon kilometer drabbades han av total utmattning och måste lägga sig för att vila, men krafterna återkom efter en stund och löpningen kunde genomföras i god fart. Han drabbades även under denna tid av magont och kräkningar och togs in på sjukhus. Ikterus konstaterades, men när leversjukdom kunde uteslutas bedömdes gulsoten som förenlig med morbus Gilbert. Efter militärtjänsten har han arbetat som telemontör och busschaufför och klarat sig bra, väl medveten om sin fysiska begränsning.
Den yngre systern har varit stillsam och uppfattats som klen sedan barndomen. Att tex åka cykel i uppförsbackar har inte varit möjligt, och vid skidtävlingar i skolan har hon kommit bland de sista. I grundskolan och i gymnasiet har gymnastiken varit ett bekymmer, då hon av lärarna betraktats som lat eller maskande. Även hon har varit lätt ikterisk så länge hon kan minnas. Hon har fött två barn utan komplikationer och hushållsarbetet har gått problemfritt, men att skjuta barnvagn i uppförsbacke upplevdes som en prövning.
Det fjärde fallet i släkten, sonen till den äldre brodern, uppfattades av föräldrarna som relativt inaktiv som barn och har i skolan undvikit fysisk träning och tävlingar. Dock har han också varit med om fullständig oförmåga att röra benen efter att ha pressat sig på cykel i en uppförsbacke. Han, liksom de övriga, uppvisar klara hemolystecken (Tabell I).
Muskelbiopsier har tagits från de tre manliga västerbottenspatienterna för histokemisk färgning. Ingen säker PFK-aktivitet har kunnat påvisas [Thornell LE, anatomiska institutionen, Umeå universitet, pers medd, 2005].
Jämtlandsfamiljen
Dottern (Figur 2, II:1) i familjen behandlades vid 13 års ålder för astmatiska symtom, som emellertid var av övergående natur. Tre år senare remitterades hon från vårdcentral till sjukhus på grund av lätt gulsot. Vid intagningen till barnkliniken konstaterades att allmäntillståndet var gott utan sjukdomstecken, ingen lever- eller mjältförstoring förlåg.
Laboratorieresultaten visade hemolysbild: högt bilirubin, ökat antal retikulocyter och en haptoglobinnivå på nära 0 g/l. Ingen anemi sågs (Tabell I). Som ett led i den fortsatta utredningen skickades blodprov till kemiska laboratoriet vid Norrlands universitetssjukhus i Umeå för osmotisk resistensbestämning. Denna friade från tankar på sfärocytos, men provet hade tagits med heparin som antikoagulans (fria Ca-joner), och efter autoinkubation över natten vid 37°C framträdde en markant ökad osmotisk resistens (se nedan). Detta påminde om fynden från Västerbottensfamiljen, varför misstankar om Taruis sjukdom väcktes.
Patienten har inte känt sig svag i musklerna men heller inte utsatt sig för kroppsansträngningar utöver det nödvändiga, tex i skolgymnastiken. Som 20-åring har hon försökt förbättra sin fysik med cykling och styrketräning; detta utan att ha drabbats av muskelsmärtor.
Hennes yngste bror (Figur 2, II:3) har undersökts på sjukhus ett flertal gånger för magsmärtor både till höger och vänster i buken. Tillståndet har uppfattats som tecken på leverinflammation. Kvarstående ikterus har föranlett hemolysutredning med bla bestämning av erytrocyternas osmotiska resistens vid kemiska laboratoriet i Umeå. Resultatet blev detsamma som för systern, ökad osmotisk resistens med Ca-joner i plasma efter autoinkubation av provet.
Den undersökningen har sedan fullföljts vid Akademiska sjukhusets kemiska laboratorium i Uppsala och har visat enzymaktivitet och metabolitförändringar i de röda blodkropparna, typiska för fosfofruktokinasbrist (Tabell II). Någon säker kraftnedsättning eller minskad uthållighet vid olika bollspel eller skidåkning har inte gått att få stöd för anamnestiskt.
Genetisk bakgrund
Cirka 15 mutationer i PFK-M-genen, associerade med Taruis sjukdom, har rapporterats från olika delar av världen [4]. Undersökning av Västerbottensfamiljen har påvisat två mutationer som inte beskrivits hos andra patienter med denna sjukdom. Den ena mutationen består av ett basutbyte (G till A) i sista nukleotiden i exon 13 (E13), den andra av ett basutbyte (A till G) i intron 16 (IVS16). Båda mutationerna förutsägs resultera i prematur avslutning av mRNA-translationen med resultatet att någon fungerande PFK-M-subenhet inte bildas [14]. De tre syskonen i Västerbottensfamiljen är sammansatt heterozygota för defektallelerna liksom sonen i tredje generationen. Denna pseudodominans förklaras av att sonens mor är anlagsbärare av mutation E13, samma som hennes svärmor, dock utan känt släktskap (Figur 1).
Undersökning av Jämtlandsfamiljen med avseende på de två mutationerna påvisar E13 i heterozygot form hos de två undersökta barnen och modern. De två barnen i denna familj antas därför vara sammansatt heterozygota för mutation E13 samt en okänd mutation (Figur 2).
De två syskonens föräldrar härstammar från Västerbottens kustområde, som tycks vara källan för PFK-mutationerna. Västerbottensfamiljens hemort är en fjällnära kommun, men sannolikt har sjukdomsanlagen förts västerut i samband med koloniseringen av övre Norrlands inland under 1700-talet.
Sammanfattning
Ronquist och medarbetare har, som nämnts ovan, upptäckt ett tidigare okänt fenomen hos de röda blodkropparna hos personer med Taruis sjukdom [7, 8]. Erytrocyterna får höjd kalciumkoncentration på grund av ökat, passivt läckage av kalciumjoner genom membranet. Den intrakorpuskulära kalciumökningen anses ge minskad deformabilitet med ökad membranviskositet hos de röda blodkropparna, med försvårad mjältpassage och hemolys som synligt resultat in vivo. Ytterligare en kalciumjoneffekt är möjlig genom att de negativa laddningarna i membranets fosfatidylserin förskjuts till korpuskelns utsida, vilket kan aktivera koagulationsprocessen [15, 16].
En ökad pool av fria kalciumjoner i erytrocyterna leder också till ett selektivt utträde av kaliumjoner efter aktivering av speciella kaliumjonkanaler. Denna specifika effekt av kalciumjonerna intracellulärt, sk Gardos-effekt, leder också till vattenutträde så att de röda blodkropparna skrumpnar (xerocytos). Detta kan registreras som en tydlig minskning av MCV och kan förklara den ökade osmotiska resistensen efter autoinkubation i heparinplasma (kalciumjoner närvarande).
Den genotyp som finns representerad i den först beskrivna familjen leder till myopati, men av mycket varierande svårighetsgrad. Orsaken till skillnaderna i fenotyp hos de drabbade är okänd. Jämtlandspatienterna har, i motsats till västerbottningarna, ingen säker ansträngningsintolerans. E13-mutationen anses vara en noll-allel, medan den andra allelen, som inte är kartlagd, möjligen kan ge en defekt M-peptidsyntes med sänkt PFK, med minskad aktivitet i de flesta fallen. Aktiviteten kan dock vara tillräcklig för normal muskelglykolys och pyruvatbildning.
Som jämförelse kan nämnas att myofosforylasaktivitet som inte underskrider 20 procent av det normala anses kunna bevara symtomfrihet hos McArdle-patienter [17]. Minskad enzymaktivitet, men över en viss kritisk gräns, kan medföra besvärsfrihet även vid ansträngning.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.

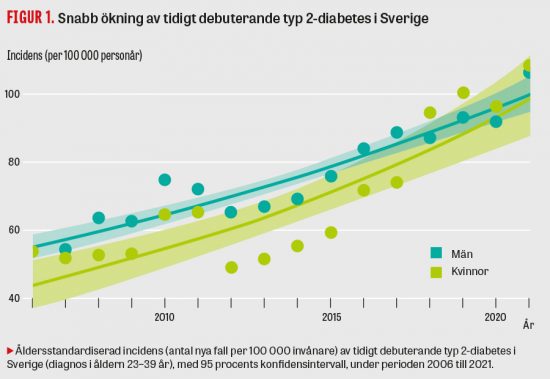
Figur 1. Västerbottensfamiljens släktträd. II:2, II:3 och II:4 är tre av tio nu levande syskon. I:3 är avliden och DNA-prov fanns inte. I:2 och I:4 är sannolikt släkt, men släktskap har inte kunnat spåras i kyrkböcker.

Figur 2. Jämtlandsfamiljens släktträd. II:2 är symtomfri och inte undersökt genetiskt.

Är tabellen svårläst hänvisar vi till nedladdningsbar pdf (högst upp på denna sida).

Är tabellen svårläst hänvisar vi till nedladdningsbar pdf (högst upp på denna sida).
Referenser
1. Valentine WN, Tanaka KR, Paglia DE. Pyruvate kinase and other enzyme deficiency disorders of the erythrocyte. In: Scriver CR, editor. Metabolic basis of inherited disease.New York: McGraw-Hill; 1989; p. 2341-65.
2. Tanaka KR, Zerez CR. Red cell enzymopathies of the glycolytic pathway. Semin Hematol 1990;27:165-85.
3. Tarui S, Okuno G, Ikura Y, Tanaka T, Suda M, Nishikawa M. Phosphofructokinase deficiency in skeletel muscle. A new type of glycogenosis. Biochem Biophys Res Commun 1965;19:517-23.
4. Nakajima H, Raben N, Hamaguchi T, Yamasaki T. Phosphofructokinase deficiency; past, present and future. Curr Molec Med 2002; 2:197-212.
5. Vora S, Davidson M, Seamen C, Miranda AF, Noble NA, Tanaka KR, et al. Heterogeneity of the molecular lesions in inherited phosphofructokinase deficiency. J Clin Invest 1983;72:1995-2006.
6. Fujii H, Miwa S. Recent progress in the molecular genetic analysis of erythro-enzymopathy. Am J
Hematol 1990;34:301-10.
7. Ronquist G, Rudolphi O, Engström I, Waldenström A. Familial phosphofructokinase deficiency is associated with a disturbed calcium homeostasis in erythrocytes. J Intern Med 2001;249:85-95.
8. Waldenström A, Engström I, Ronquist G. Increased erythrocyte content of Ca in patients with Tarui
disease. J Intern Med 2001;249: 97- 102.
9. Haller RG, Vissing J. No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology 2004;62:82-6.
10. Löfberg M, Jänkälä H, Paetau A, Härkönen M, Somer H. Metabolic causes of recurrent rhabdomyolysis. Acta Neurol Scand 1998;98: 268-75.
11. Exantus J, Ranchin B, Dubourg L, Touraine R, Baverel G, Cochat P. Acute renal failure in a patient with phosphofructokinase deficiency. Pediatr Nephrol 2004;19:111-3.
12. Vissing J, Haller RG. A diagnostic cycle test for McArdle disease. Ann Neurol 2003;54:539-42.
13. Rudolphi O, Ek B, Ronquist G. Inherited phosphofructokinase deficiency associated with hemolysis and exertional myopathy. Eur J Haematol 1995;55:279-81.
14. Nichols RC, Rudolphi O, Ek B, Exelbert R, Plotz PH, Raben N. Glycogenosis type VII (Tarui disease) in a Swedish family: two novel mutations in muscle phosphofructokinase gene (PFK-M) resulting in intron retentions. Am J Human
Genet 1996;59:59-65.
15. Finsterer J, Stöllberger C, Kopsa W. Neurologic and cardiac progression of glycogenosis type VII over an eight-year period. South Med J 2002;95:1436-40.
16. Ronquist G. Glycogenosis type VII (Tarui disease): diagnostic considerations and late sequelae. South Med J 2002;95:1361-2.
17. El-Schahawi M, Tsujino S, Shanske S, DiMauro S. Diagnosis of McArdle disease by molecular genetic analysis of blood. Neurology 1996;47: 579-80.
Summary
SUMMARY
We have previously reported on a family of four members with Tarui disease in the county of Västerbotten in Sweden. Now we have identified another two cases, a brother and a sister, with hemolysis but with no clear myopathy due to deficiency of phosphofructokinase of M-type. Probably the two families are related as they share one mutation, the other one in the Jämtland-family is not yet investigated. The erythrocytes from the four patients of the first family were shown to have an augmented membrane leakage of Ca-ions and as a result an elevated intracellular Ca-ion pool. The concomitant hemolysis may be explained by a diminished erythrocyte deformability due to Ca-ion overload.
OLLE RUDOLPHI, GÖRAN BRATTSAND, ANDERS WALDENSTRÖM,
ANNA-LENA NILSSON,GUNNAR RONQUIST.
Correspondence: Gunnar Ronquist, Department of Medical Sciences, Clinical
Chemistry, Akademiska sjukhuset, SE-751 85 Uppsala, SWEDEN.
(Gunnar.Ronquist@akademiska.se)