Sammanfattat
Det nyupptäckta peptidhormonet hepcidin är involverat i den störda järnomsättningen vid såväl inflammation som hereditär hemokromatos.
Hepcidin bildas i levern och blockerar järnupptaget i tarmen samt järnfrisättningen från makrofagerna, vilket leder till sänkt serumjärn.
Vid inflammation ses höga hepcidinnivåer, eftersom hepcidinsyntesen induceras av interleukin-6.
Järn inducerar hepcidinsyntesen via cellytemolekylerna HFE, hemojuvelin och transferrinreceptor2. Mutationer i någon av dessa leder till hepcidinbrist och hereditär hemokromatos. Vanligast är mutationer i HFE.
Det järnöverskott som ses vid kronisk leversjukdom och alkoholöverkonsumtion anses bero på minskad hepcidinsyntes i levern. Järnöverskott förekommer också vid insulinresistens, men där är orsaken okänd.
Vid obesitas kan fettväven bilda hepcidin, med sänkt serumjärn som följd.
I mitten av 1800-talet rapporterades att man vid obduktion av patienter med triaden levercirros, ökad hudpigmentering och diabetes kunde finna ökad järninlagring i parenkymatösa organ. Sjukdomen betraktades som en raritet. Den beskrevs mer i detalj 1889 av von Recklinghausen, som myntade begreppet bronsdiabetes [1]. Det dröjde ända till 1935 innan sjukdomen beskrevs systematiskt av Sheldon, som benämnde den hemokromatos [2].
På 1950-talet introducerades venesektion, blodtappning, som en effektiv behandling mot järnöverskottet. Under 1960-talet uppstod en debatt om huruvida sjukdomen var ärftlig eller associerad till alkoholöverkonsumtion. Debatten tystnade när man konstaterade att det finns en koppling till vissa HLA-typer, främst HLA A3 och B7, och att hemokromatosgenen kunde lokaliseras till korta armen på kromosom 6 [3]. Det fastställdes att sjukdomen ärvs autosomalt recessivt.
Nästa genombrott kom 1996 då Feder och medarbetare kunde visa att 90 procent av patienter med hereditär hemokromatos var homozygota för en mutation i en gen som då benämndes HLA-H [4]. Något år senare döptes genen om till HFE (det märkliga är att ingen säkert vet vad denna bokstavskombination står för). Man kunde visa att HFE-proteinet uttrycks på cellytan, binder till transferrinreceptorn och reglerar upptaget av järn till cellen, men den bakomliggande mekanismen var oklar [5].
År 1999 upptäcktes en andra transferrinreceptor (TfR2) specifik för leverceller och ytterligare ett transportprotein för järn, kallat ferroportin. Man fann att mutationer i dessa gener också leder till hemokromatos [6, 7].
År 2001 upptäcktes den antimikrobiella peptiden hepcidin som blockerar järnupptaget i tarmen [8] och att mutationer i dess gen leder till en ovanlig form av juvenil hemokromatos [9]. År 2004 konstaterades att juvenil hemokromatos också orsakas av mutationer i en gen som kodar för ett annat cellyteprotein, som döptes till hemojuvelin (HJV) [10].
Inom loppet av åtta år hade man således upptäckt fem nya gener involverade i järnomsättningen, vilka kunde vara muterade vid hemokromatos, där HFE dock är den absolut dominerande.
Man introducerade en klassifikation av hemokromatos baserad på de olika mutationerna [11] (Tabell I). Denna klassifikation har fått kritik, eftersom det har visat sig att det inte föreligger någon strikt korrelation mellan genotyp och fenotyp. Man har tex funnit patienter med den juvenila formen av hemokromatos som har dubbelmutationer i HFE och TfR2 [12] och andra patienter med mutationer i HJV som har den klassiska adulta och inte den juvenila formen av hemokromatos [13].
Därför föreslogs nyligen en klassifikation som baseras på klinisk bild, där begreppen »adult« och »juvenil« hereditär hemokromatos introduceras [12]. Ett förslag till en sådan klassifikation anpassad efter svensk terminologi presenteras i Fakta 1.
Adult hereditär hemokromatos – oftast mutationer i HFE
Den vanligaste orsaken till hereditär hemokromatos är mutationer i HFE-genen, vilket påträffas hos 90 procent av patienterna i norra Europa [14]. Homozygoti för utbytet av cystein till tyrosin i aminosyraposition 282 (C282Y) i HFE-proteinet är den dominerande mutationen. C282Y påträffas i heterozygot form hos 7 procent av den vita befolkningen i norra Europa, medan homozygoti för C282Y ses hos 0,3–0,5 procent [15]. Mutationen anses ha uppstått hos en keltisk förfader för 60–70 generationer sedan (år 0–500 eKr) [16]. Sannolikt har den utgjort en fördel för överlevnad genom att reducera risken för anemi under graviditet, och den har därför anrikats kraftigt i befolkningen.
Enbart heterozygoti för C282Y, utan andra ärftliga faktorer som påverkar järnomsättningen, anses inte leda till järnöverskott utan snarare till god järnbalans i övre delen av normalområdet. Hos patienter med homozygoti för C282Y ses biokemiskt järnöverskott med förhöjt S-ferritin och/eller S-järnmättnad hos 70–80 procent av männen och 50–70 procent av kvinnorna [17-19].
Det finns alltså ett relativt stort antal individer som bär på mutationen i homozygot form men som ändå har helt normala järnparametrar. De faktorer som påverkar penetransen är manligt kön [20] och graden av alkoholkonsumtion [21]. Man har inte kunnat visa att dieten i övrigt har någon avgörande betydelse för sjukdomsutveckling. Dessutom finns andra genetiska faktorer, hittills okända, som påverkar penetransen.
Den andra mutationen, H63D (utbyte av histidin mot asparaginsyra på aminosyra 63) leder i sig inte till hemokromatos, inte ens i homozygot form. Denna mutation är spridd i olika befolkningsgrupper och ses hos cirka 15 procent av befolkningen [15]. Det är endast i kombination med C282Y, sk sammansatt heterozygoti, som den kan leda till järnöverskott. Emellertid utvecklar endast 20 procent av individer med sammansatt heterozygoti förhöjd järnmättnad och 34 procent förhöjd ferritinnivå [18].
HFE-associerad hemokromatos är ofta lindrig
Kliniken vid hereditär hemokromatos inkluderar skrumplever, diabetes, hypogonadism, artros, hjärtsvikt, arytmier och risk för hepatocellulär cancer [22, 23], vilket visar att flera organsystem kan engageras. Dessa kliniska fenotyper härrör från fall med höga ferritinvärden; patienter som sökt sjukvård på grund av sina symtom och där diagnosen uppdagats efter utredningen.
I genanalysens tidevarv träder en annan bild fram. I en populationsstudie av Adams och medarbetare genotypades 100000 individer varpå 299 med homozygoti för C282Y upptäcktes [17]. Man fann ingen skillnad i frekvensen av artrit, diabetes, hjärtsjukdom, infertilitet eller impotens mellan dem med homozygoti och dem som saknade HFE-mutationer. Däremot sågs ökad frekvens leversjukdom hos dem med homozygoti, illustrerat av bla förhöjda leverenzymvärden. Frekvensen av leverfibros hos patienter med homozygoti för C282Y förefaller ligga mellan 4 och 19 procent, och levercirros ses hos 1,5–7 procent [18, 19]. I en studie är också ledbesvär (18 procent) mer frekventa bland individer med homozygoti än bland befolkningen i övrigt (5 procent) [24].
Dessa data utesluter inte det faktum att enstaka patienter med mycket höga ferritinvärden kan utveckla såväl diabetes som hypogonadism och hjärtbesvär till följd av sitt järnöverskott, men den stora majoriteten individer med homozygoti för C282Y förblir friska livet ut.
Utredning och behandling av järnöverskott
Diagnostik och behandling av järnöverskott visas schematiskt i Fakta 2. Utredningen av misstänkt hemokromatos inkluderar kontroll av S-ferritin och S-järnmättnad vid minst två tillfällen, fP-glukos, S-ALAT, S-ALP, B-blodstatus och HFE-mutationsanalys [25]. Man har inte kunnat påvisa järnrelaterade leverskador vid ferritinvärden <1000 (my)g/l [26]. Om HFE-mutationsanalys påvisar genotypen C282Y/C282Y (homozygoti) eller C282Y/H63D (sammansatt heterozygoti) och S-ferritin är förhöjt men fortfarande <1000 (my)g/l, kan behandling påbörjas utan att leverbiopsi behöver göras. Om S-ferritin >1000 (my)g/l rekommenderas emellertid leverbiopsi.
Vid misstanke om cirros görs också leverbiopsi, ultraljudsundersökning av levern och gastroskopi. Vid misstanke om hjärtsjukdom utförs EKG och ekokardiografi. Om patienten saknar ovanstående mutationer i HFE men misstanken om hemokromatos kvarstår, skall diagnostisk leverbiopsi övervägas.
Behandlingen utgörs av venesektion, 400–500 ml per vecka, tills S-ferritin är kring 50 (my)g/l. Patienten tolererar denna intensivbehandling väl. Man kan acceptera att B-hemoglobin sjunker till nedre normalvärdesgränsen. Om det blir lägre kan man överväga kontroll av S-kobalamin och S-folat.
Efter avslutad intensivbehandling övergår man till underhållsbehandling med 2–6 blodtappningar per år. Intervallet styrs av årliga kontroller av S-ferritin, där man eftersträvar ett värde kring 50 (my)g/l. Vissa blodcentraler accepterar patienter i denna fas som blodgivare.
Eventuella ledbesvär behandlas med antiflogistika. Tyvärr förbättras sällan artropatibesvären av venesektion. Eftersom det föreligger ökad risk för hepatocellulär cancer hos patienter som utvecklat cirros, även efter framgångsrik blodtappning, väljer många centra att följa dessa patienter med cirros med ultraljudskontroller exempelvis var sjätte månad [23].
Syskon till patienter med HFE-associerad hemokromatos bör genotypas, eftersom de har 25 procents sannolikhet att ha ärvt samma genotyp [25].
HFE reglerar syntesen av järnhormonet hepcidin
Alltsedan HFE-proteinets upptäckt har man försökt kartlägga mekanismerna för dess reglering av järnbalansen. En central landvinning gjordes 2001 då man hos mus karakteriserade en cysteinrik peptid, vars expression ökade vid dietärt järnöverskott [27]. Peptiden benämndes hepcidin. Därefter har det publicerats över 200 artiklar om detta järnreglerande hormon, vars centrala roll för järnhomeostasen nu har klarnat.
Hög järnmättnad stimulerar syntesen av hepcidin i levern. HFE är en av flera molekyler som reglerar denna syntes i leverns hepatocyter. Två andra är TfR2 och HJV. Hepcidin syntetiseras som en 84 aminosyror lång prepropeptid, vilken spjälkas till den aktiva peptiden innehållande 25 aminosyror (benämnd hepcidin-25) [28].
Hos människa har hepcidin-25 påträffats både i blod och i urin [29, 30]. Strukturellt uppvisar hepcidin-25 stora likheter med andra antimikrobiella peptider, bla i form av fyra disulfidbryggor. Dessutom har hepcidin-25 antimikrobiell och antifungal effekt in vitro. Huruvida hepcidin-25 har antimikrobiella egenskaper också in vivo är ännu inte klarlagt.
Hepcidinets huvudsakliga roll rör dock inte dess antimikrobiella egenskaper, utan dess kraftfulla effekter på järnhomeostasen i kroppen.
Hepcidin hämmar järnfrisättningen till plasma
Efter det att peptiden utsöndrats från hepatocyterna till plasma utövar den sin verkan på makrofager och tarmceller (främst i den proximala delen av tunntarmen) (Figur 1). Den binder till en specifik receptor, ferroportin, som är ett transmembranöst protein [31]. Ferroportin återfinns i det basala cellmembranet i enterocyterna, i retikuloendoteliala celler, i leverns hepatocyter och i placentas syncytiotrofoblaster.
Ferroportin är ett transportprotein som exporterar järn från cellen till plasma. När hepcidinet binder till ferroportin internaliseras det sistnämnda i cellen och degraderas [32]. Järnet blir då kvar intracellulärt, och järnmättnaden i serum sjunker. I knockoutmöss där hepcidingenen slagits ut ses ett kraftigt järnöverskott i lever och parenkymatösa organ [8]. Mutationer i hepcidinets aktiva del har beskrivits hos patienter med juvenil hereditär hemokromatos [33]. Hepcidinmutationer som orsak till hereditär hemokromatos är emellertid extremt sällsynta.
Brist på hepcidin leder till hemokromatos
I experiment på cellkulturer har HFE-proteinet på cellens yta visats interagera med transferrinreceptor1 (TfR1), vilket reglerar det transferrinmedierade järnupptaget till cellen. Mutationen C282Y resulterar i att HFE-proteinet aldrig når cellytan [31], vilket leder till sänkt hepcidinsyntes, även om den inte är helt utsläckt [34]. Låga hepcidinnivåer i serum leder till minskad degradering av ferroportin. Ökad ferroportinaktivitet resulterar i att enterocyter och makrofager snabbt tömmer sitt innehåll av intracellulärt järn till plasma. Järnmättnaden stiger samtidigt som makrofager och enterocyter blir järnfattiga (Figur 2).
Förekomsten av järnfattiga makrofager i benmärg har länge ansetts typisk för hereditär hemokromatos [25] och har differentierat denna från sekundär hemokromatos, där man ser ökad mängd järn i benmärg och i makrofager (även i leverns Kupffer-celler). Den höga järnmättnaden leder till sakta progredierande järnöverskott främst i levern, typiskt för adult hereditär hemokromatos.
Hereditär hemokromatos kan också bero på mutationer i TfR2
Transferrinreceptor2 (TfR2) identifierades 1999, och ett år senare upptäcktes att en italiensk släkt med HFE-negativ hemokromatos hade mutationer i denna receptor [6]. TfR2 skiljer sig från TfR1 genom att ha en svagare bindning till transferrinbundet järn och genom att receptorantalet på cellytan inte minskar vid järnöverskott. Dessutom verkar TfR2 direkt kunna reglera syntesen av hepcidin, eftersom TfR2-knockoutmöss, liksom patienter med mutationer i TfR2, har försämrat hepcidinsvar på järn och får järninlagring i parenkymatösa organ [35].
Mutationer i TfR2 är mer frekventa i Medelhavsområdet. Vi undersökte förekomsten av denna mutation hos 44 svenska hemokromatospatienter som saknade mutationer i HFE, men kunde inte finna ett enda fall [15]. Nyligen har det beskrivits fall med en kombination av mutationer i HFE och TfR2, vilket ledde till mycket låga hepcidinnivåer och utveckling av juvenil hemokromatos [12]. Mutationsanalys av TfR2 används för närvarande inte i klinisk praxis.
Mutationer i HJV kan leda till juvenil hemokromatos
En annan regulator av hepcidinsyntesen är hemojuvelin (HJV). HJV upptäcktes 2004 [10], och dess funktion är okänd. Proteinet uttrycks i lever, tarm, skelettmuskel, hjärta, hjärna och njurar [36]. Mycket talar för att det har en central roll i järnomsättningen i flera av kroppens organ, eftersom HJV-knockoutmöss utvecklar kraftig järninlagring i parenkymatösa organ [37].
Hos människa leder homozygoti för vissa mutationer i HJV till kraftigt nedsatt hepcidinsvar på järn. Dessa patienter får uttalad hepcidinbrist och mycket kraftig järninlagring (juvenil hemokromatos) [10]. De kan få symtom på hjärtsvikt, leverskador och diabetes redan i 20-årsåldern. Lyckligtvis är juvenil hemokromatos extremt sällsynt, med endast ett 30-tal fall beskrivna i litteraturen. Nyligen har det publicerats ett antal fall, där andra mutationer i HJV gett upphov till en lindrigare klinisk bild, som mer liknar adult hemokromatos [13].
»Ferroportin disease« – ovanlig form av hemokromatos
I genen för järnexportören ferroportin finns ett stort antal mutationer beskrivna [7, 38]. Dessa kan leda till syntes av ett defekt protein som aggregerar i det endoplasmatiska retiklet och inte når cellytan. Detta resulterar i minskad järntransport till plasma, ackumulation av järn i makrofager, enterocyter och hepatocyter samt låg järnmättnad i serum.
Denna fenotyp kallas »ferroportin disease«, och i motsats till hemokromatos, som nedärvs autosomalt recessivt, ärvs »ferroportin disease« autosomalt dominant. Detta beror på att syntesen av det normala ferroportinet (från den icke-muterade allelen) hämmas av aggregationen av defekt protein i det endoplasmatiska retiklet.
Kliniskt kännetecknas »ferroportin disease« av kraftigt förhöjt ferritin men normal (eller låg) järnmättnad i serum [38]. B-Hb-värdet är normalt eller lätt sänkt. Leverbiopsi visar järninlagring i främst makrofager (Kupffer-celler). Patienter med »ferroportin disease« har sämre tolerans för venesektion, eftersom Hb och serumjärn lätt sjunker efter blodtappning. Ibland får man därför ge adjuvant behandling med erytropoietin [11].
»Ferroportin disease« har beskrivits främst i Frankrike och är en ovanlig form av hemokromatos; förekomsten av denna form av järninlagringssjukdom har ännu inte undersökts i Sverige.
Alkohol och kronisk leversjukdom leder till sänkt hepcidin
I klinisk praxis har man länge förundrats över den järninlagring i levern som kan ses vid kronisk leversjukdom, cirros och excessivt alkoholintag, utan att det föreligger hemokromatos. Sannolikt beror denna sideros på minskad syntes av hepcidin. De facto har det nyligen visats att den ökade oxidativa stress som ses vid metabolismen av etanol leder till minskad hepcidinsyntes [39, 40]. På samma sätt sjunker hepcidinsyntesen i levern vid kronisk leverskada, exempelvis vid cirros eller kronisk hepatit C, vilket kan förklara den sideros i levern som kan ses vid dessa tillstånd [41, 42].
Järnöverskott kan vara associerat med insulinresistens
Vid insulinresistens och metabolt syndrom ses ofta lätt till måttligt förhöjda ferritinvärden, lindrig järninlagring i levern, men normal järnmättnad i serum. Denna typ av »metabolt järnöverskott« (insulin resistance-associated hepatic iron overload, IR-HIO) beskrevs 1999, och den anses vara 10 gånger vanligare än hereditär hemokromatos [43]. Dessa patienter har inga påvisbara mutationer i HFE, och orsaken till den ökade järninlagringen är okänd. Hepcidinnivåerna hos denna patientkategori har inte studerats, och ett eventuellt samband mellan glukosomsättning och hepcidinsyntes i levern är ännu så länge spekulativt. Dessa patienter får sällan ferritinnivåer över 1000 (my)g/l.
Histologiskt är siderosen ofta lindrig. Ibland föreligger samtidig steatohepatit (icke-alkoholorsakad steatohepatit, NASH [non-alcoholic steatohepatitis]), där ökad järnmängd kan bidra till leverpåverkan [44].
Patienter med metabolt järnöverskott i levern svarar på venesektion med sjunkande ferritin- och transaminasvärden [45], och i en studie har man även visat att venesektion förbättrar insulinkänsligheten [46]. Ofta görs därför venesektion på dessa patienter, fast med glesare intervall än vad som är praxis vid hemokromatos.
För mycket hepcidin leder till anemi
Förhöjda hepcidinnivåer ses vid vissa ovanliga tumörformer och vid inflammationer och leder till låg järnmättnad i plasma (Figur 3). Leveradenom som överproducerar hepcidin leder till terapirefraktär anemi som hävs först då tumören avlägsnas [47]. Det är känt sedan länge att vid inflammatoriska tillstånd ses järnbrist, anemi och förhöjda ferritinnivåer. Anemin vid kronisk sjukdom är normokrom och kan inte hävas med peroral järntillförsel, eftersom järnupptaget i tarmen är blockerat av den höga hepcidinaktiviteten. Denna typ av anemi undanhåller järn från bakterier vid infektiösa tillstånd och hämmar på så sätt infektionens förlopp [28, 48]. Vid icke-infektiösa inflammatoriska tillstånd som reumatoid artrit eller inflammatorisk tarmsjukdom är anemin dock ett icke önskvärt tillstånd som i stället bidrar till sjukdomssymtomen.
Interleukin-6 inducerar hepcidinsyntesen i levern
Man har nu visat att interleukin-6 (IL-6), som frisätts vid kronisk inflammation, är den mest kraftfulla stimulatorn av hepcidinsyntesen [49]. IL-6 leder till stigande hepcidinnivåer i plasma, varvid järnet ackumuleras i makrofagerna, järnupptaget i tarmen stängs av och järnmättnaden i serum sjunker. De ökade järndepåerna i retikuloendoteliala celler leder till stigande nivåer S-ferritin.
Det har visats att patienter med inflammatoriskt medierad anemi har höga hepcidinnivåer i urinen [42]. Vid inflammatoriska tillstånd som medieras av IL-6 kan således det låga serumjärnet, det höga ferritinet, den normokroma anemin och den dåliga effekten av peroralt järn samtliga tillskrivas ökad hepcidinaktivitet.
Den behandling som står till buds idag vid dessa tillstånd är intravenöst järn och erytropoietin. Intravenöst järn förefaller inte aggravera den inflammatoriska processen vid exempelvis inflammatorisk tarmsjukdom på samma sätt som perorala järnpreparat kan göra [50]. Emellertid skulle eventuell framtida behandling med hepcidinantagonister kunna reversera denna typ av anemi, som är relaterad till kronisk inflammation.
I en nyligen publicerad studie har man också påvisat hepcidinsyntes i fettväven hos patienter med svår obesitas. Mängden hepcidin-mRNA i fettväven korrelerade till inflammationsparametrar som C-reaktivt protein och IL-6, men var inte relaterad till förekomst av diabetes eller NASH i levern. Dessa patienter hade låg järnmättnad i serum, och hos vissa sågs också järnbristanemi [51].
Ännu finns ingen tillförlitlig metod att mäta hepcidin
De studier som gjorts på hepcidin har påvisat reglering av hepcidinsyntesen i levern på mRNA-nivå. Det har utvecklats en urinanalys som finns beskriven i forskningssammmanhang men som inte används i rutinbruk. Halter av hepcidin i urinen korrelerar till mRNA-nivåerna i levern hos människa [42]. Tyvärr finns det ännu inte någon tillförlitlig hepcidinanalys för mätning i serum. Mutationsanalys av TfR2, HJV, hepcidin eller ferroportin används inte heller ännu i klinisk rutin.
Upptäckten av järnhormonet hepcidin har givit nya spännande perspektiv på järnhomeostasen och dess reglering. Såväl brist som överskott på hepcidin eller defekter i receptorn ferroportin leder till störd järnomsättning och kan leda till sjukdomsutveckling. I ett framtidsperspektiv kan analys av hepcidinnivåer i serum och behandling med hepcidinagonister och -antagonister erbjuda nya diagnostiska och terapeutiska möjligheter vid handläggningen av patienter med hemokromatos, inflammationsmedierad anemi och metabolt järnöverskott.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
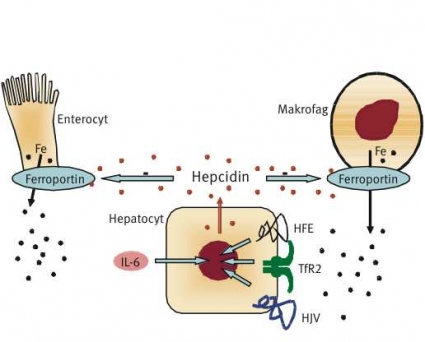
Figur 1. Normal järnbalans och avsaknad av inflammation. Nivåerna av interleukin-6 (IL-6) är låga och påverkar inte hepcidinsyntesen. Mängden transferrinbundet järn i serum reglerar hepcidinsyntesen via transferrinreceptor2 (TfR2), HFE och HJV (hemojuvelin). Hepcidinnivåerna stiger vid god tillgång på järn och sjunker vid järnbrist. Hepcidinet binder till ferroportin lokaliserat i det basolaterala membranet i enterocyter och makrofager. Hepcidin leder till att ferroportinet degraderas och järnupptaget minskar. (Vid järnbrist sker det motsatta; låga hepcidinnivåer leder till ökad järntransport via ferroportin och stigande serumjärn.)
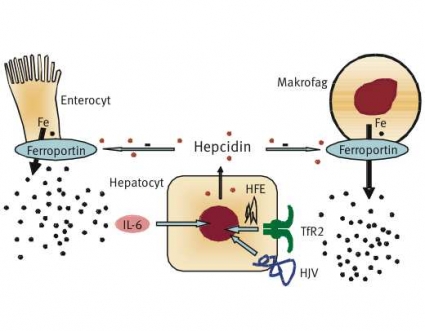
Figur 2. Järnbalans vid hereditär hemokromatos. (Bilden illustrerar mutation i HFE-genen, vilken leder till ett defekt protein som inte når cellytan.) Mutationer som påverkar funktionen av transferrinreceptor2 (TfR2), HFE och/eller hemojuvelin (HJV) leder till upphävd reglering av hepcidinsyntesen och oproportionerligt låga hepcidinnivåer i plasma. Hepcidinets hämmande effekt på järnupptaget (via degradering av ferroportin) minskar. Järnupptaget ökar. Enterocyter och makrofager blir järnfattiga, eftersom det järn som transporteras in i dessa celler (hos enterocyten via tarmlumen, hos makrofagen via fagocytos av röda blodkroppar) snabbt pumpas ut i serum via ferroportin. Järnmättnaden ökar i serum, vilket leder till progredierande järninlagring i parenkymatösa organ och stigande ferritinnivåer.
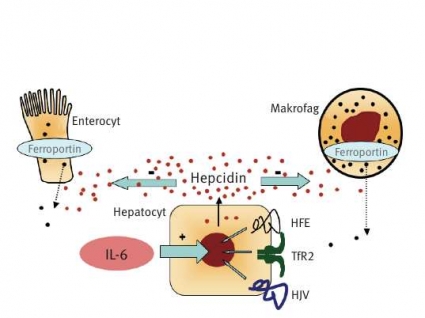
Figur 3. Järnbalans vid inflammation. Höga nivåer av interleukin-6 (IL-6) inducerar hepcidinsyntesen. Höga hepcidinnivåer i plasma leder till degradation av ferroportin och minskad transport av järn från enterocyter och makrofager till serum. Järn retineras i makrofager och enterocyter. Järnmättnaden i serum minskar. Retentionen av järn i makrofager leder till stigande S-ferritinnivåer. (TfR2=transferrinreceptor2, HJV=hemojuvelin.)

Om tabellen är svårläst hänvisar vi till nedladdningsbar pdf (högst upp på denna sida).
Referenser
1. von Recklinghausen FD. Über hemokromatose. Berlin Klinische Wochenschrift 1889, 26: 925-935.
2. Sheldon JH. Hemochromatosis. Lancet 1934, Nov 10: 1031-1036.
3. Simon M, Bourel M, Genetet B, Fauchet R. Idiopathic hemochromatosis. Demonstration of recessive transmission and early detection by family HLA typing. N Engl J Med 1977; 297: 1017-1021.
4. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399-408.
5. Waheed A, Parkkila S, Zhou XY, Tomatsu S, Tsuchihashi Z, Feder JN, et al. Hereditary hemochromatosis: effects of C282Y and H63D mutations on association with beta2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci U S A. 1997;94:12384-9.
6. Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25:14-5.
7. Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, et al. Autosomal dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001;108:619-23.
8. Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98:8780-5.
9. Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21-2.
10. Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77-82.
11. Franchini M. Hereditary iron overload: update on pathophysiology, diagnosis, and treatment. Am J Hematol. 2006;81:202-9.
12. Pietrangelo A, Caleffi A, Henrion J, Ferrara F, Corradini E, Kulaksiz H, et al. Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology. 2005;128:470-9.
13. Lee PL, Barton JC, Brandhagen D, Beutler E. Hemojuvelin (HJV) mutations in persons of European, African-American and Asian ancestry with adult onset haemochromatosis. Br J Haematol. 2004;127:224-9.
14. Cardoso EM, Stål P, Hagen K, Cabeda JM, Esin S, de Sousa M, Hultcrantz R. HFE mutations in patients with hereditary haemochromatosis in Sweden. J Intern Med. 1998;243:203-8.
15. Holmström P, Marmur J, Eggertsen G, Gåfvels M, Stål P. Mild iron overload in patients carrying the HFE S65C gene mutation: a retrospective study in patients with suspected iron overload and healthy controls. Gut. 2002;51:723-30.
16. Lucotte G, Dieterlen F. A European allele map of the C282Y mutation of hemochromatosis: Celtic versus Viking origin of the mutation? Blood Cells Mol Dis. 2003;31:262-7.
17. Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769-78.
18. Olynyk JK, Cullen DJ, Aquilia S, Rossi E, Summerville L, Powell LW. A population based study of the clinical expression of the hemochromatosis gene. N Engl J Med. 1999;341:718-24.
19. Åsberg A, Hveem K, Thorstensen K, Ellekjter E, Kannelonning K, Fjosne U, et al. Screening for hemochromatosis: high prevalence and low morbidity in an unselected population of 65,238 persons. Scand J Gastroenterol. 2001;36:1108-15.
20. McCune CA, Ravine D, Carter K, Jackson HA, Hutton D, Hedderich J, et al. Iron loading and morbidity among relatives of HFE C282Y homozygotes identified either by population genetic testing or presenting as patients. Gut. 2006;55:554-62.
21. Scotet V, Merour MC, Mercier AY, Chanu B, Le Faou T, Raguenes O, et al. Hereditary hemochromatosis: effect of excessive alcohol consumption on disease expression in patients homozygous for the C282Y mutation. Am J Epidemiol. 2003;158:129-34.
22. Niederau C, Fischer R, Purschel A, Stremmel W, Häussinger D, Strohmeyer G. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 1996; 110: 1107-1119.
23. Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:13-20.
24. Deugnier Y, Jouanolle AM, Chaperon J, Moirand R, Pithois C, Meyer JF, et al. Gender-specific phenotypic expression and screening strategies in C282Y-linked haemochromatosis: a study of 9396 French people. Br J Haematol. 2002;118:1170-8.
25. Stål P, Hagen K, Hultcrantz R. Trots nya genupptäckter: störningen vid hemokromatos är fortfarande oklar. Läkartidningen. 1998;95:3430-5.
26. Olsson KS, Ritter B, Lundin PM. Liver affection in iron overload studied with serum ferritin and serum aminotransferases. Acta Med Scand. 1985;217:79-84.
27. Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin,
is overexpressed during iron overload. J Biol Chem. 2001;276:7811-9.
28. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003 Aug 1;102(3):783-8.
29. Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial Activity. FEBS Lett. 2000;480:147-50.
30. Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001 Mar 16;276(11):7806-10.
31. Anderson GJ, Frazer DM. Hepatic iron metabolism. Semin Liver Dis. 2005;25:420-32.
32. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090-3.
33. Matthes T, Aguilar-Martinez P, Pizzi-Bosman L, Darbellay R, Rubbia-Brandt L, Giostra E, et al. Severe hemochromatosis in a Portuguese family associated with a new mutation in the 5\'-UTR of the HAMP gene. Blood. 2004;104:2181-3.
34. Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, et al. Disrupted
hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361(9358):669-73.
35. Wallace DF, Summerville L, Lusby PE, Subramaniam VN. First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut. 2005;54:980-6.
36. Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005;115:2180-6.
37. Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187-91.
38. Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131-8.
39. Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281:22974-82.
40. Milward E, Johnstone D, Trinder D, Ramm G, Olynyk J. The nexus of iron and inflammation in hepcidin regulation: SMADs, STATs, and ECSIT. Hepatology. 2007;45:253-6.
41. Nagashima M, Kudo M, Chung H, Ishikawa E, Hagiwara S, Nakatani T, Dote K.
Regulatory failure of serum prohepcidin levels in patients with hepatitis C. Hepatol Res. 2006;36:288-93.
42. Detivaud L, Nemeth E, Boudjema K, Turlin B, Troadec MB, Leroyer P, et al. Hepcidin levels in humans are correlated with hepatic iron stores, haemoglobin levels, and hepatic function. Blood. 2005;106:746-8.
43. Mendler MH, Turlin B, Moirand R, Jouanolle AM, Sapey T, Guyader D, et al. Insulin resistance-associated hepatic iron overload. Gastroenterology. 1999;117:1155-63.
44. Bugianesi E, Manzini P, D\'Antico S, Vanni E, Longo F, Leone N, et al. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in non-alcoholic fatty liver. Hepatology. 2004;39:179-87.
45. Guillygomarc\'h A, Mendler MH, Moirand R, Laine F, Quentin V, David V, et al. Venesection therapy of insulin resistance-associated hepatic iron overload. J Hepatol. 2001 Sep;35(3):344-9
46. Fernandez-Real JM, Penarroja G, Castro A, Garcia-Bragado F, Hernandez-Aguado I, Ricart W. Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and betacell function. Diabetes. 2002;51:1000-4.
47. Chung A, Leo K, Wong G, Chuah K, Ren J, Lee C. Giant hepatocellular adenoma presenting with chronic iron deficiency anemia. Am J Gastroenterol. 2006;101:2160-2.
48. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461-3.
49. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271-6.
50. Erichsen K, Ulvik RJ, Nysaeter G, Johansen J, Ostborg J, Berstad A, Berge RK,
Hausken T. Oral ferrous fumarate or intravenous iron sucrose for patients with inflammatory bowel disease. Scand J Gastroenterol. 2005;40:1058-65.
51. Bekri S, Gual P, Anty R, Luciani N, Dahman M, Ramesh B, et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology. 2006;131:788-96.