Sammanfattat
Fas I-prövningar är viktiga humanfarmakologiska studier med syfte att kartlägga säkerhet, tolerabilitet, farmakokinetik och/eller farmakodynamik hos ett läkemedel.
Efter att samtliga sex friska försökspersoner som erhöll den monoklonala antikroppen TGN 1412 (en biologisk substans) i en första-gången-till-människa(FIM)-studie drabbats av livshotande biverkningar har fokus riktats mot säkerhetsaspekter i tidiga fas I-prövningar.
Av 149 fas I-prövningar som åren 1995, 2000 och 2005 genomförts i Sverige med substanser som ännu inte godkänts för försäljning var det tre som avslogs och sju som avbröts av säkerhetsskäl.
Av de tio prövningar som avslogs/avbröts av säkerhetsskäl var fyra FIM-studier och sex andra fas I-studier innan substansen godkänts för försäljning.
Biologiska substanser var överrepresenterade bland prövningar med säkerhetsproblem jämfört med kemiska substanser.
Trots den relativt omfattande säkerhetsvärdering som sker på myndigheter i dag inträffade den 13 mars 2006 en läkemedelskatastrof med en ny biologisk substans, TGN 1412, som testades för första gången på människa. Samtliga sex friska försökspersoner i London som erhöll försöksläkemedlet drabbades av akuta livshotande biverkningar i form av cytokinfrisättningssyndrom [1, 2]. Sedan dess har säkerheten i tidiga fas I-prövningar livligt debatterats [se tex 3-7], vilket bla har resulterat i att ett nytt europeiskt riktlinjedokument har tagits fram [8].
Med syftet att få bättre underlag för att kunna avgöra om TGN 1412-katastrofen utifrån olika aspekter var en enskild företeelse med liten risk att upprepas och för att öka kunskapen om utvecklingen av fas I-prövningar i Sverige gjordes en genomgång av samtliga ansökningar om fas I-prövningar till Läkemedelsverket under åren 1995, 2000 och 2005.
Mot bakgrund av att TGN 1412 är en biologisk substans och med beaktande av påtagliga skillnader mellan biologiska och kemiska substanser avseende egenskaper som immunogenicitet och farmakokinetik valde vi att studera biologiska och kemiska substanser separat. Med biologisk substans avses här en substans som tillverkats från ett biologiskt ursprungsmaterial eller med hjälp av en biologisk metod såsom rekombinant teknik.
Med fas I-prövningar avses humanfarmakologiska studier med syfte att kartlägga säkerhet, tolerabilitet, farmakokinetik och/eller farmakodynamik.
Metod
Sedan 1984 måste alla kliniska läkemedelsprövningar i Sverige godkännas av Läkemedelsverket. Ansökningarna granskas av ett flertal olika personalkategorier inkluderande bla prekliniker/toxikolog, läkare, statistiker och farmaceut. Protokollet och annan prövningsrelaterad information finns sparad i ett manuellt arkiv på Läkemedelsverket. Prövningarna registreras även sedan 1988 i en databas.
Inom ramen för projektet identifierades samtliga fas I-prövningar under åren 1995, 2000 och 2005. Prövningsansökningarna studerades med avseende på bla typ av substans (biologisk eller kemisk), hur tidigt i utvecklingen av substansen studien genomfördes (första-gången-till-människa, FIM, annan studie innan substansen godkänts för försäljning till människa eller studie efter godkännandet), hur väl protokollet angav på vilket sätt och i vilken tidsordning substansen skulle administreras, studiens design, val av patientgrupp, Läkemedelsverkets värdering av prövningen och förekomst av säkerhetsproblem.
Begreppet säkerhetsproblem definierades som preklinisk eller klinisk säkerhetsinformation som ledde till att prövningen inte godkändes eller till att den avbröts tillfälligt eller permanent. Sedan den 1 maj 2004 läggs samtliga kliniska prövningar i EU in i en gemensam databas, EudraCT. För år 2005 kunde vi därför jämföra förekomsten av fas I-prövningar i Sverige gentemot övriga länder i EU.
Genom att söka i tillgänglig dokumentation i form av Läkemedelsverkets databas över registrerade produkter och arkivet för kliniska prövningar, vid behov kompletterat med aktiv kontakt med läkemedelsföretag och/eller sökning på Internet, försökte vi även ta reda på huruvida substanser som ännu inte var godkända senare blivit godkända eller lagts ner.
Resultat
Totalt identifierades 263 fas I-prövningar, varav tre (1 procent) inte återfanns i arkivet och därför inte inkluderades i projektet.
Fas I-prövningarnas fördelning
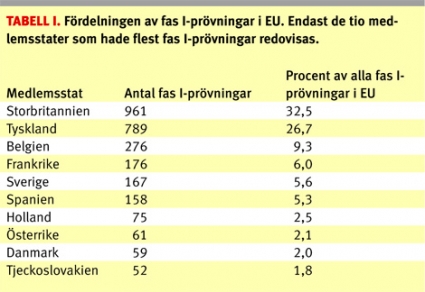
Figur 1 visar en översikt över det årliga antalet fas I-prövningar i relation till det totala antalet prövningar i Sverige sedan datasystemet för kliniska prövningar på Läkemedelsverket infördes. Man kan notera en stadig nedgång i antalet kliniska prövningar i Sverige sedan registreringen startade som dock tycks ha stannat upp under de senaste åren. Andelen fas I-prövningar inom EU år 2005 framgår av Tabell 1, där Storbritannien och Tyskland stod för en majoritet av prövningarna, medan Sverige hamnade på femte plats.
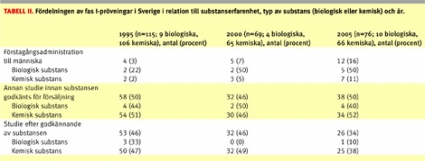
Av Tabell II framgår fördelningen av prövningarna i relation till substanserfarenhet, typ av substans (biologisk eller kemisk) och år. Man kan notera en ökning av FIM-prövningar över tiden för såväl biologiska som kemiska substanser. Fördelningen av prövningar på substanserfarenhet skiljde sig dock mellan biologiska och kemiska substanser med en tydlig förskjutning mot FIM-prövningar för biologiska substanser.
Studiedesign och studiepopulation
Ökad användning av placebo noterades: från 14 procent (16/ 115) år 1995 till 30 procent (21/69) år 2000 och 39 procent (30/74) år 2005. Patienter förekom oftare i FIM-prövningar än i övriga fas I-prövningar (48 procent; 10/21 mot 18 procent; 42/239). Andelen studier som inkluderade kvinnor var något lägre år 1995 (53 procent; 61/115) än år 2000 (71 procent; 59/69) eller år 2005 (66 procent; 50/74).
I FIM-prövningarna ökade andelen studier som inkluderade kvinnor från 25 procent (1/4) år 1995 till 40 procent (2/5) år 2000 och 67 procent (8/12) år 2005. Det noterades även en ökning av antalet försökspersoner åren 2000 och 2005 (medianvärden 18 respektive 24) jämfört med år 1995 (medianvärde 12) för prövningar med substanser som ännu inte godkänts för försäljning.
Administration och säkerhet
Information om hur väl protokollet angav på vilket sätt och i vilken tidsordning substansen skulle administreras inhämtades för prövningar som genomfördes med substanser som ännu inte godkänts för försäljning (n=149). Endast fem protokoll (3 procent; 5/149) hade tydligt angivit på vilket sätt och när i förhållande till nästa individ administreringen skulle ske. Av dessa rörde endast ett protokoll en studie med en substans som skulle administreras för första gången till människa (5 procent; 1/21 sådana prövningar).
Av de 149 prövningarna med substanser som ännu inte godkänts för försäljning var det totalt tre (2 procent) som inte godkändes på grund av säkerhetsproblem och 8 (5 procent) som avbröts på grund av säkerhetsproblem. Två av tre prövningar som inte godkändes rörde FIM-studier med biologiska substanser. I båda fallen bedömdes det prekliniska underlaget vara otillräckligt, och de data som fanns indikerade möjliga problem. Den tredje prövningen rörde en annan tidig fas I-studie, dock ej FIM, med en kemisk substans där nytillkomna prekliniska studier uppvisade oacceptabla toxikologiska fynd som bedömdes kräva mekanistisk kartläggning innan ytterligare studier på människa skulle kunna accepteras.
De säkerhetsproblem som låg till grund för att åtta prövningar avbröts rörde i tre fall prekliniska toxikologiska fynd (kemiska substanser; ingen FIM), i tre fall biverkningsproblem i studier på människa (två kemiska och en biologisk substans; ingen FIM) och i ett fall vardera teoretisk risk för mutagenicitet med användning av retrovirala vektorer (biologisk substans; FIM) respektive identifiering av en genotoxisk metabolit (kemisk substans; FIM).
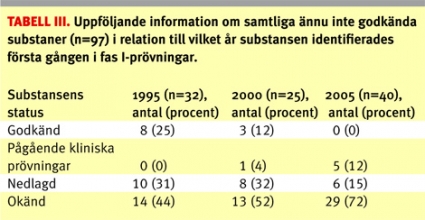
Följande biverkningsproblem noterades: en oroväckande hög frekvens av neutropeni ledde till att en kemisk substans lades ned, ett fall med allvarlig psykisk biverkning i en studie med en kemisk substans ledde till att studien avbröts tillfälligt och ett flertal fall avseende neutropeni utanför Sverige med en biologisk substans ledde till att studien avbröts i Sverige. Den totala andelen prövningar med säkerhetsproblem var högre för biologiska (21 procent; 4/19) än för kemiska (5 procent; 7/130) substanser. De 149 prövningarna som genomfördes på substanser som ännu inte godkänts för försäljning avsåg totalt 97 olika substanser. För en överblick över andelen substanser som senare godkänts eller lagts ner, se Tabell III.
Diskussion
Vissa trender över tid noterades beträffande den statistiska designen och inklusionen av kvinnor. Användning av placebo blev vanligare och antalet studerade försökspersoner/patienter ökade, vilket kan tala för en utveckling mot tydligare karakterisering av effekt och biverkningar tidigt i det kliniska prövningsprogrammet. Introduktion av placebo medger jämförelse med obehandlade, och den därtill blindade designen torde minska förekomsten av störfaktorer som kan uppkomma som ett resultat av kännedom om vad som administrerats.
Kvinnoinklusion och fördelning inom EU
Det faktum att kvinnoinklusion blivit vanligare över tid återspeglar sannolikt en strävan att tidigt i det kliniska prövningsprogrammet inkludera individer som är representativa för den tilltänkta patientpopulationen. Efter talidomidkatastrofen ansågs det länge oetiskt att exponera kvinnor i fas I-studier med tanke på eventuella risker i samband med graviditet. Med tiden har dock synen på kvinnoinklusion blivit mer balanserad, då det är av betydelse att få information även från kvinnor. Risken för graviditet kan dessutom hanteras genom att exempelvis endast kvinnor som inte är fertila inkluderas.
Det är intressant att se på vilket sätt fas I-prövningar fördelar sig över EU-medlemsländerna. Sverige var det femte största landet för fas I-prövningar år 2005, vilket är en bra placering med tanke på folkmängden. Storbritannien och Tyskland intog emellertid en särställning, vilket i Storbritannien kan hänga samman med det faktum att fas I-prövningar inte behövde godkännas av läkemedelsmyndigheten innan det EU-gemensamma kliniska prövningsdirektivet trädde i kraft.
Substansernas öde
Vi fann att 25 procent av substanser som ännu inte godkänts och identifierats bland fas I-prövningar år 1995 så småningom godkändes. Siffran är lägre för år 2000 (12 procent), men det är möjligt att denna siffra kan justeras upp i framtiden. När det gäller år 2005 är det ännu för tidigt att studera godkännandefrekvensen. Det skulle vara av värde med ett säkrare system för att kunna följa upp substansernas öde, då mörkertalet är relativt stort.
Biologiska substanser med större säkerhetsproblem
Biologiska substanser var behäftade med en större andel säkerhetsproblem än kemiska substanser i denna studie. De biologiska substanserna omfattar vitt skilda typer av produkter, exempelvis vacciner (som kan innehålla genetiskt material med eller utan vektorer i form av plasmider eller virus), celler med särskilda egenskaper, som produktion av ett visst protein eller antigenpresenterande celler, monoklonala antikroppar, proteinkedjor, peptider etc. Man kan spekulera över om biologiska substanser i större utsträckning testas på mekanismer vars effekter hittills är okända. Detta kan leda inte bara till ökad misslyckandefrekvens i klinisk forskning utan också till svårigheter att demonstrera verkningsmekanismens relevans i djurmodeller. Denna studie var emellertid för liten för att kunna uttala sig närmare om detta.
Oklarheter kring läkemedelsadministration
Det är oundvikligt att administration av läkemedel till människa är förenat med risker. Det är därför viktigt att alla som deltar i klinisk prövningsverksamhet är väl införstådda med vilka eventuella risker som kan förekomma, att dessa i största möjliga mån förebyggs och att man har beredskap att ta hand om dem. Vår studie visade att en detaljerad redovisning i protokollet, där det framgår på vilket sätt och när i förhållande till nästa individ administrationen av substansen skulle äga rum, generellt sett var en ovanlig företeelse under åren 1995–2005. Endast en FIM-prövning hade en sådan detaljerad redogörelse.
Konklusion
Som väntat identifierades dock inga säkerhetsproblem av det slag som inträffade vid TGN 1412-katastrofen, och den totala förekomsten av säkerhetsproblem var låg. Dock var det relativt få FIM-prövningar. Som ett resultat av TGN 1412-katastrofen har Läkemedelsverket infört ännu striktare säkerhetsrutiner. En tydlig redovisning i protokollet av hur lång tid som måste passera från det att substansen administrerats till den första personen i studien till dess att den får administreras till följande person(er) är i princip obligatorisk för FIM-prövningar. Krav ställs även på att dos och administrationssätt är väl motiverade från säkerhetssynpunkt så att personerna i studien inte utsätts för onödiga risker.
För substanser som bedöms kunna medföra säkerhetsrisker men samtidigt stora potentiella terapeutiska vinster ökar kravet på att inkludera patienter i stället för friska försökspersoner. Sannolikheten för en upprepning av TGN 1412-katastrofen torde vara liten oavsett vilka ytterligare säkerhetsåtgärder som vidtas men är om möjligt ännu mindre som ett resultat av de nya säkerhetsrutinerna.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
*
Hans Melander var idégivare och drog upp riktlinjerna för studien.
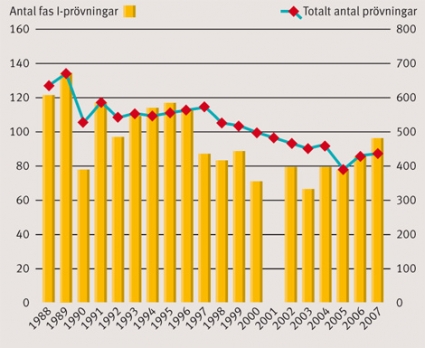
Figur 1. Antal fas I-prövningar per år i relation till det totala antalet prövningar i Sverige åren 1988–2007. För år 2001 saknas uppgift om antalet fas I-prövningar.
Om tabellen är svårläst hänvisar vi till nedladdningsbar pdf (högst upp eller längst ner på denna sida).
Referenser
1. Marshall E. Lessons from a failed drug trial. Science. 2006;313:901.
2. Wadman M. London´s disastrous drug trial has serious side effects for research. Nature. 2006;440: 388-9.
3. Goodyear M. Learning from the TGN1412 trial. BMJ. 2006;332: 677-8.
4. Mayor S. Severe adverse reactions call for trial design changes. BMJ. 2006;332:683.
5. Mayor S. Inquiry into adverse events in trial blames drug, not study design. BMJ. 2006;332:870.
6. Day M. Agency critisizes drug trial. BMJ. 2006;332:1290.
7. Lafolie P. Varför blev läkemedelsprövningen vid Northwick Park Hospital en katastrof? Läkartidningen. 2006;103:1169-70.
8. Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products. Committee for medicinal products for human use. Doc. Ref. EMEA/CHMP/SWP/28367/07.