I Läkartidningen nr 3/2009 publicerades två artiklar om aktuell diagnostik och behandling av polycythaemia vera (PV) och essentiell trombocytemi (ET) [1]. Nu vill vi belysa primär myelofibros, som är den tredje sjukdomen i denna grupp kal- lad kroniska myeloproliferativa sjukdomar, MPD (eller, med senaste benämnningen, kroniska myeloproliferativa neoplasier, MPN). Vi diskuterar här viktiga nyheter inom diagnostik och klassifikation samt de nya behandlingsmöjligheter som är på väg och som ger hopp om bättre omhändertagande av patienter med myelofibros. Myelofibros har av hävd ansetts vara den svåraste av de tre sjukdomarna, utan bra behandling och med kort överlevnad. Att så inte är fallet för alla är en viktig ny information.
Primär myelofibros uppfattas oftast som mindre vanlig än PV och ET, men sporadiska populationsbaserade studier har rapporterat stora skillnader i incidens. Enligt en ny klassifikation kommer många fall som tidigare klassificerats som ET att nu i stället klassificeras som primär myelofibros. Det är inte svårt att gissa att detta leder till en ökning av antalet fall av myelofibros men även till att många patienter kan uppfattas ha en betydligt allvarligare sjukdom än ET. Det är därför viktigt att vi kan förmedla kunskap om sjukdomens heterogena karaktär till våra kolleger och till patienter som får diagnosen primär myelofibros.
Terminologi
Vid primär myelofibros saknar patienterna tecken på föregående blodsjukdom. Hälften av dem har en mutation i JAK2-genen, samma mutation som vid PV. Med noggrann anamnes och grävande i gamla journaler kan man dock ofta spåra en tyst, icke-diagnostiserad PV (eller ET) hos många. Patienterna kan ha haft den för PV så typiska akvatiska klådan, eller så har de fått höra att de har haft »fina blodvärden«, något som vid senare analys visar sig vara ett förhöjt hemoglobinvärde, inte dramatiskt förhöjt men klart ovanför referensvärdena. Dessa fall benämns ibland post-PV- (eller post-ET)-myelofibros. Kliniskt skiljer sig inte dessa patienter från dem med primär myelofibros, och vi gör i denna artikel fortsättningsvis inte skillnad på dem.
Fibros i benmärgen ses någon gång som del av en annan sjukdom, t ex systemisk skleros, Sjögrens sjukdom, primär biliär cirros [2], infektionssjukdomar, pulmonell hypertension [3] eller cancermetastaser. Vid vissa former av myelodysplastiskt syndrom och lymfom ses också benmärgsfibros, som kan leda till differentialdiagnostiska resonemang.
Incidens
I en studie från Göteborgsområdet diagnostiserades åren 1983–1999 totalt 43 fall av primär myelofibros, varav 20 män och 14 kvinnor i åldrarna 49–90 år. I denna studie beräknades incidensen till 0,3 per 100 000 invånare och år [4]. Sjukdomen är vanligast hos äldre men förekommer även sällsynt hos unga och barn. Som nämnts ses myelofibros även som slutstadium till PV och ET, som ofta behandlas enligt samma principer som gäller för primär myelofibros.
Klinisk bild och diagnostiska kriterier
Primär myelofibros karakteriseras av benmärgsfibros, förekomst av omogna vita blodkroppar i perifert blod och splenomegali. Den senare kan vara uttalad. Blodutstryket uppvisar en karakteristisk bild, med representation av flertalet förstadier till neutrofiler och avvikande morfologi hos erytrocyter. Typiskt är fyndet av tårformade erytrocyter (dakrocyter). Typiskt är också anemi och förhöjt laktatdehydrogenas, som ingår i de diagnostiska kriterierna (Fakta 1). Antalet trombocyter och leukocyter kan vara kraftigt förhöjt vid diagnostillfället, men med progredierande sjukdom brukar det sjunka, och speciellt grav trombocytopeni kan bli ett uttalat problem i slutstadiet av sjukdomen.
Begreppet benmärgsfibros är således centralt och innebär att blodbildande benmärg ersätts av bindväv, främst retikulin och kollagenfibrer. Samtidigt sker dock en dramatisk ökning av antalet blodkärl i benmärgen [5]. Denna intensiva angiogenes har lockat till funderingar om huruvida den är en motsvarighet till tumörformande maligniteters täta men oorganiserade kärlnätverk, som är nödvändigt för att försörja tumören med syre och näring. Det är även möjligt att se den intensiva kärlnybildningen vid myelofibros som ett sätt att försöka motverka, läka, en skada.
Prognos och riskbedömning
Primär myelofibros har betydligt sämre prognos än PV och ET. Medianöverlevnaden anges till 4–5,5 år. Vanliga dödsorsaker är transformation till akut leukemi, infektioner, trombos eller blödning och komplikationer till portal hypertension [6]. Flera studier har dock kunnat visa att en grupp patienter med primär myelofibros har betydligt bättre prognos, och för att identifiera dem har ett flertal prognostiska system konstruerats genom åren [7, 8].
Nyligen publicerades resultatet av ett stort internationellt samarbete, där data från över 1 000 nydiagnostiserade patienter från sju olika centra användes för att identifiera de starkaste prognostiska faktorerna vid det tillfälle då diagnosen primär myelofibros ställdes. Fem faktorer som innebär hög risk identifierades (Fakta 2). Detta poängsystem kallas International Prognostic Scoring System (IPSS) och identifierar fyra riskgrupper med en medianöverlevnad som varierar mellan 27 och 135 månader. IPSS utgör således en god grund för att bedöma förväntad prognos och kan vara till hjälp vid terapival, t ex vid bedömning om patienten bör genomgå allogen benmärgstransplantation [9].
Genetiska förändringar vid primär myelofibros
Mutation i JAK2-genen, V617F, som beskrevs 2005, förekommer hos ungefär hälften av patienterna med primär myelofibros och kan utgöra en diagnostisk hjälp för att särskilja sjukdomen från andra sjukdomar med benmärgsfibros [10]. JAK2-mutationen förekommer även vid ET och PV och hos så gott som alla patienter med post-PV-myelofibros. Vid primär myelofibros har en funktionellt liknande mutation hittats i trombopoetinreceptorgenen MPL, och denna har rapporterats förekomma hos 5 procent av patienterna. Nyligen har också mutationer i TET2-genen på kromosom 4q24 beskrivits hos knappt 15 procent av patienterna med både JAK2-muterad och -omuterad myeloproliferativ sjukdom [11, 12].
Cytogenetiska aberrationer kan påvisas hos ca 30 procent av patienterna med primär myelofibros [4]. I fall där diagnosen inte är klar kan påvisande av cytogenetiska aberrationer vara till diagnostisk hjälp, men i IPSS utföll dock inte cytogenetik som en oberoende prognostisk variabel. Helt nyligen publicerades data från Mayokliniken som visar att cytogenetik kan ge additiv prognostisk information om man exkluderar de cytogenetiska aberrationer som indikerar bättre prognos, nämligen 13q– och 20q– och bara studerar övriga klonala avvikelser [13]. JAK2-mutation gav däremot ingen prognostisk information i denna studie.
Prefibrotisk myelofibros
Ett signum för primär myelofibros är ökningen av bindväv i benmärgen. Benmärgsfibros kan dock förekomma vid ett flertal andra tillstånd, som tidigare nämnts. Att korrekt ställa diagnosen primär myelofibros är ofta komplicerat, och moderna diagnoskriterier är baserade inte bara på benmärgsmorfologi utan på hela den kliniska presentationen av sjukdomen. Likaså varierar graden av fibros vid primär myelofibros: från diskret retikelökning till manifest osteoskleros. Man uppfattar i dag att benmärgsförändringarna vid primär myelofibros utvecklas gradvis och att det även finns ett prefibrotiskt stadium där benmärgsfibrosen ännu är obefintlig eller diskret.
Begreppet prefibrotisk myelofibros infördes i WHO-klassifikationen redan 2001 och återkommer i den nya upplagan från 2008 [14]. Prefibrotisk myelofibros definierar ett tidigt stadium av sjukdomen utan eller med mycket lätt retikulinfibros i benmärgen. Diagnosen är en utmaning för patologen och baseras på morfologi och distribution av megakaryocyter. Kliniskt är dessa patienters sjukdom ofta i en proliferativ fas, med högt antal trombocyter, normalt eller lätt förhöjt leukocytantal och lätt anemi. I tidigare klassifikationer, som baserats på kriterier från Polycythemia Vera Study Group (PVSG), fick dessa patienter diagnosen ET. Patienter med prefibrotisk myelofibros anses ha betydligt större risk att senare utveckla manifest myelofibros än de patienter med förhöjda trombocyter som i dag får diagnosen ET.
Det är dock viktigt att påpeka att bra prospektiva studier som visar detta ännu saknas. Det är också viktigt att betona att flertalet av patienterna som får diagnosen prefibrotisk myelofibros har betydligt bättre förväntad överlevnad än vad konventionella överlevnadssiffror för primär myelofibros visar. Behandlingen av prefibrotisk myelofibros bör rimligen inriktas på att motverka progress av myelofibros och övergång till avancerad primär myelofibros [13].
Begreppet prefibrotisk myelofibros är kontroversiellt och har resulterat i omfattande diskussioner. I en engelsk retrospektiv studie, där tre erfarna hematopatologer blindat fick ställa diagnos enligt WHO-klassifikationen på 370 patienter med ET enligt gamla kriterier baserade på PVSG, visades reproducerbarheten mellan patologerna vara dålig. I gruppen patienter som fått diagnosen prefibrotisk myelofibros var det ingen vars sjukdom progredierade till manifest primär myelofibros under uppföljningstiden på 68 månader [15]. Begreppet prefibrotisk myelofibros kommer dock att finnas kvar även i den nya WHO-klassifikationen från 2008, och det är viktigt att öka kunskapen bland patologer i Sverige kring denna diagnostik.
Konventionell behandling
Behandlingsmöjligheterna vid myelofibros har länge varit mycket begränsade och huvudsakligen inriktats på att lindra symtomen. Fortfarande saknas tydlig evidens för att farmakologisk terapi kan ändra sjukdomens progress. I sjukdomens proliferativa fas syftar behandlingen till att minska de tromboemboliska komplikationerna. Sporadiska data antyder att cytoreduktion i detta skede kan minska fibrosprogressen, men här saknas prospektiva studier [16, 17]. I första hand rekommenderas hydroxiurea och till enstaka patienter busulfan. Interferon kan vara ett alternativ, speciellt till yngre patienter, och detta är intressant eftersom det finns flera sporadiska rapporter om fibrosregress efter tidigt insatt interferonbehandling [18].
I senare faser av sjukdomen kan cytopenier och uttalade konstitutionella symtom vara huvudproblem. Erytropoetin kan minska anemin, speciellt i de fall där serumerytropoetin ligger under 125 U/l [19, 20]. Talidomid i kombination med steroider [21] eller anabola steroider [19] kan vara ett alternativ. Splenektomi innebär betydande peroperativa risker – och komplikationer hos mer än 25 procent av opererade patienter har rapporterats [22] – men kan innebära lindring för patienter med besvär av en förstorad mjälte och ibland också för patienter med cytopenier. Det nordiska vårdprogrammet för MPD, som kan hittas via ‹http://www.sfhem.se>, ger en utmärkt översikt över konventionella behandlingsalternativ vid primär myelofibros.
Benmärgstransplantation
Den enda botande behandling som finns i dag vid primär myelofibros är allogen benmärgstransplantation. Tidigare studier av myeloablativ benmärgstransplantation har dock visat betydande mortalitet, speciellt hos patienter över 45 år. Under senare år har dock flera grupper, inklusive den svenska MPD-gruppen, publicerat studier av icke-myeloablativ transplantation vid primär och sekundär myelofibros med betydligt bättre sjukdomsfri överlevnad och lägre terapiinducerad mortalitet.
Icke-myeloblativ transplantation har kunnat genomföras framgångsrikt på äldre patienter, upp mot 65–70 år, där andra komplicerande sjukdomar inte förelegat. Resultaten har varit lika bra med obesläktad som med besläktad stamcellsdonator. I den svenska studien identifierades 27 patienter som transplanterats 1982–2004, varav tio genomgått icke-myeloablativ transplantation. Nio av tio patienter var långtidsöverlevande och uppvisade gradvis reversering av benmärgsfibrosen och mjältförstoringen samt i det närmaste en normalisering av blodvärdena [23]. I det nordiska vårdprogrammet rekommenderas att överväga benmärgstransplantation på högriskpatienter upp mot 65 års ålder med primär myelofibros. Nyligen publicerades en retrospektiv genomgång från tre stora centra för MPD som visade att icke-transplanterade patienter under 60 år inte hade sämre överlevnad än transplanterade. Fortfarande saknas randomiserade studier som visar värdet av transplantation vid myelofibros, och i väntan på dessa finns inga data som ger grund för att rekommendera transplantation till lågriskpatienter [24].
JAK2-inhibitorer
Upptäckten av en specifik mutation i JAK2-genen V617F hos mer än 95 procent av patienterna med PV och ca 50 procent av patienterna med ET och primär myelofibros har skapat nya möjligheter för diagnostik och behandling av MPD. Mutationen i JAK2-genen resulterar i en autonom överaktivitet av tyrosinkinaset Jak-2, som är en viktig länk i signaleringen från receptorerna för erytropoetin, trombopoetin och den granulocytkolonistimulerande faktorn. Intresset är således stort när data från nya JAK2-inhibitorer börjar komma. Ännu finns inga publicerade data, men uppgifter från prekliniska och kliniska studier har presenterats vid flera internationella möten.
Det preparat som kommit längst kallas INCB18424. Studier pågår även i Sverige. Preparatet är en oral JAK2-inhibitor som också har betydande effekt på Jak-1 och andra tyrosinkinaser. In vitro inhiberar läkemedlet bildningen av cytokinoberoende erytroida kolonier från patienter med PV. Preparatet eliminerar neoplastiska celler och ger förlängd överlevnad i försöksdjursmodeller med JAK2 V617F. I humana studier har preparatet visats ha en dramatisk effekt på förstorad mjälte, och patienter med betydande splenomegali har rapporterats få en snabb och bestående minskning eller normalisering av mjältens storlek. Effekten kan uppfattas som farmakologisk splenektomi. Sätts preparatet ut växer mjälten snabbt. Läkemedlet har också en dramatisk effekt på s k B-symtom, där framför allt kakexi kan vara ett stort problem för patienten. Den dosbegränsande toxiciteten visar sig, trots effekten på mjältstorleken, vara trombocytopeni.
TG10134 är en oralt tillgänglig selektiv JAK2-inhibitor, som utvecklas av TargeGen, San Diego, USA. På ett möte i Berlin rapporterades nyligen data från en fas 1-studie från Mayokliniken i USA. Även detta preparat tycks minska mjältstorleken och B-symtomen men också reducera andelen omogna celler i perifert blod. Det som gör detta preparat extra intressant är att studien också visade reduktion av andelen celler med JAK2 V617F. Om detta också innebär en påverkan på progressen av primär myelofibros återstår dock att visa.
CEP-701 är en inhibitor av FLT3, som också inhiberar både muterad och omuterad JAK2. Prövningar pågår på primär myelofibros men även på PV och ET [25].
Epigenetiska substanser
Epigenetisk modifiering av geners aktivitet har visat sig vara en viktig mekanism för många hematologiska maligniteters utveckling och progress. Vi vet föga om metyleringen av gener vid primär myelofibros, men mekanismen har visats förekomma hos enstaka patienter. Hypometylerande läkemedel såsom 5-azacytidin, som har visat mycket intressanta effekter vid myelodysplastiska syndrom, har tyvärr visat mer måttlig aktivitet vid primär myelofibros. Däremot kan histondeacetylasinhibitorer vara intressanta eftersom histondeacetylasaktiviteten har visats vara ökad vid primär myelofibros [26], och studier av dessa substanser pågår. I Sverige planeras en studie där vorinostat (suberoylanilide hydroxamic acid, SAHA) kommer att prövas kliniskt på patienter med PV eller ET.
Kvalitetsregistrering
Populationsbaserad kunskap om epidemiologin och prognosen vid myeloproliferativa sjukdomar är fortfarande ganska begränsad. I det nationella blodcancerregistret som startade 2007 ingår sedan 1 januari 2008 även myeloproliferativa sjukdomar inklusive primär myelofibros. Blodcancerregistret sköts av landets åtta onkologiska centra. Registret för MPD har utformats av den svenska gruppen, och en styrgrupp för registret med representanter för samtliga regioner har bildats. Registret ger möjlighet att studera incidensen av MPD i Sverige och att identifiera patienter för uppföljningar och kliniska studier. Det kan också ge insyn i hur diagnostiken av MPD går till runt om i landet, vilket är viktigt, eftersom den ibland kan vara komplicerad.
För att registreringen ska få framgång är det viktigt att kunskap om registret sprids till alla enheter där myeloproliferativa sjukdomar diagnostiseras i Sverige. Samtliga patienter diagnostiserade efter 1 januari 2008 ska registreras i första hand elektroniskt i IT-plattformen, som sköts av Informationsnätverket för cancervården (INCA), men formulär kan också skickas in till regionala onkologiska centra, och blanketter kan fås via varje centrums webbplats. Varje klinik har möjlighet att se sin egen registrering i Blodcancerregistret, och nationella sammanställningar av data planeras vartannat år. Vi tror att detta register ska kunna ge unik och värdefull kunskap om epidemiologin vid primär myelofibros och ge möjlighet för bättre rekrytering av patienter till kliniska studier.
Orsaker till myelofibros
Avslutningsvis vill vi ge några synpunkter på hur myelofibros uppstår. I vår tidigare artikel i Läkartidningen [1] redogjorde vi för genetiskt modifierade möss som utvecklar myelofibros. Det finns (minst) tre djurmodeller transgena för JAK2 V617F som uppvisar en klassisk PV-fenotyp. Med tiden utvecklar djuren även alla klassiska tecken på myelofibros, vilket bekräftar att mutationen kan ge upphov till myelofibros. Vad är det då som triggar igång fibrosutvecklingen och angiogenesen och får blodbildande CD34-positiva celler att lossa från benmärgen och försöka starta blodbildning i mjälten (och sannolikt i andra organ)? Vi vet inte svaret, men några mekanismer träder fram tydligare än andra.
Den första frågan är om fibros i benmärg är annorlunda än fibros i lever eller lunga. Om inte, varför förekommer då inte lung- och leverfibros oftare parallellt med myelofibros? Ett intressant fynd är att megakaryocyter kan påträffas i lungcirkulationen [2, 3, 27]. Kan vi lära av kunskapsutvecklingen i fråga om lungfibros för att bättre förstå myelofibros? Är abnorm fibros i ärrvävnad, som vid keloid, besläktad med myelofibros i sina mekanismer? Här finns gott om frågetecken och få svar.
Den andra frågan rör mediatorerna för fibrosutveckling. Det är alldeles klart att överproduktion av trombopoetin i en av djurmodellerna för MPD räcker för att skapa myelofibros. Om det är trombopoetinet självt eller om myelofibrosen uppkommer via sekundära reaktioner, t ex inverkan på andra tillväxtfaktorer, är oklart. Intressant är att myelofibros setts i enstaka fall vid behandling med de nya trombopoetinliknande substanserna, som nu finns för behandling av immunologisk trombocytopen purpura (IIP). Störst intresse har ägnats transforming growth factor-β (TGFβ) och platelet derived growth factors (PDGF) som klassiska mediatorer för bindvävsnybildning [27]. Men inget definitivt, eller så sjukligt att man kan rikta specifik behandling mot det, har hittills upptäckts i deras signalsystem vid myelofibros. Så vi står fortfarande med en rad frågetecken.
*
Potentiella bindningar eller jävsförhållanden: Mats Merup och Jan Palmblad har deltagit i advisory board för Incyte. Mats Merup är nationell koordinator för studie från Incyte.
*
Birgitta Sander, docent och överläkare, institutionen för laboratoriemedicin, Karolinska universitetssjukhuset, Huddinge, har bidragit med bild och text.


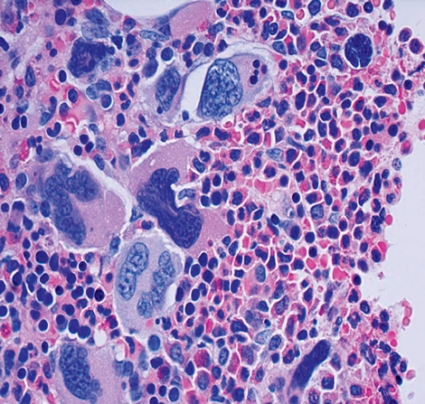
Figur 1. Benmärg från patient med prefibrotisk myelofibros. Man ser flera megakaryocyer som ligger i ett kluster eller till och med rygg mot rygg, vilket är ett patologiskt fynd. Till skillnad från normala megakaryocyter och megakaryocyter vid essentiell trombocytopeni, som uppvisar djupt loberade kärnor, har megakaryocyterna på bilden kärnor med få eller inga lobuli. De är hypoloberade/monoloberade och har beskrivits som »molnlika«. Vissa av megakaryocyterna har också hyperkromatiska, mörka kärnor. Denna typ av dysplasi associeras främst med primär myelofibros. Foto: Birgitta Sander
Referenser
1. Merup M, Löfvenberg E, Palmblad J: Nya rekommendationer om kroniska myeloproliferativa sjukdomar – Ny kunskap måste ge ny praxis. Läkartidn 2009:106:98-9.
2. Pullarkat V, Bass RD, Gong JZ, Feinstein DI, Brynes RK. Primary autoimmune myelofibrosis: definition of a distinct clinicopathologic syndrome. Am J Hematol 2003;72:8-12.
3. Popat U, Frost A, Liu E, Guan Y, Durette A, Reddy V, Prchal JT. High levels of circulating CD34 cells, dacrocytes, clonal hematopoiesis, and JAK2 mutation differentiate myelofibrosis with myeloid metaplasia from secondary myelofibrosis associated with pulmonary hypertension. Blood 2006; 107(9):3486-8
4. P. Johansson, J Kutti, B Andreasson, S Safai-Kutti, L Vilén , H Wedel, B Ridell. Trends in the incidence of chronic Philadelphia chromosome negative (Ph-) myeloproliferative disorders in the city of Göteborg, Sweden, during 1983-99. J Int6 Med 2004;, 256:, 161-65.
5. Zetterberg E, Vannucchi AM, Migliaccio RM, Vainchenker W,, Tulliez M, Dickie, R, Rogers R, Palmblad J. Pericyte coverage of abnormal vessels in myelofibrosis bone marrows. Haematologica 2007;92:597-604.
6. F Cervantes1, F Passamonti, G Barosi. Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukemia (2008:) 22:, 905-914.;
7. Dupriez B, Morel P, DemoryJL, Lai JL, SimonM, PlantierI, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood 1996;88:1013-8.
8. Cervantes F, Pereira A, Esteve J, Rafel M, Cobo F, Rozman C, et al. Identication of “short-lived” and “long-lived” patients at presentation of idiopathic myelobrosis. Br J Haematol 1997;97:635-40.
9. Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, Mesa RA, Demory JL, Barosi G, Rumi E, Tefferi A. A new prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2008; 2008;113(13):2895-901. Epub 2008 Nov 6.
10. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144-48.
11. Delhommeau F, Dupont S, James C, Masse A, le Couedic JP, Valle VD et al. TET2 is a novel tumor suppressor gene inactivated in myeloproliferative neoplasms: identification of a pre-JAK2 G617F event. ASH Annu Meet Abstr 2008; 112:Iba-Ib3. Late-breaking abstract.
12. Tefferi A, Pardanani A, Lim K-H, Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S, Mullally A, Li C-Y, Hanson CA, Mesa R, Bernard O, Delhommeau F, Vainchenker W, Gilliland DG, Levine RL. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009 E-publ ahead of print Leukemia 2009 E-publ ahead of print.
13. Hussein K, Huang J, Lasho T, Pardanani A, Mesa F, Williamson C, Ketterling R, Hanson C, Van Dyke D, Tefferi A. Karyotype complements the International Prognostic Scoring System for primary myelofibrosis. Eur J Haematol. 2009 Apr;82(4):255-9. Epub 2008 Feb 10.
14. Swedlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. WHO Press 2008.
15. Wilkins BS, Erber WN, Bareford D, Buck G, Wheatley K, East CL et al. Bone marrow pathology in essential thrombocythemia: interobserver reliability and utility for identifying disease subtypes. Blood 2008;111:60-70.
16. Löfvenberg E, Wahlin A, Roos G, Ost A. Reversal of myelofibrosis by hydroxyurea. J Haematol 1990;44:33-8.
17. Thiele J, Kvasnicka HM, Schmitt-Graeff A, Spohr M, Diehl V, Zankovich R, Niederle N, Leder LD. Effects of interferon and hydroxyurea on bone marrow fibrosis in chronic myelogenous leukaemia: a comparative retrospective multicentre histological and clinical study. Br J Haematol. 2000;108:64-71.
18. Retter A, Radia DH, Harrisson CN. Improvement of fibrosis in a patient with chronic myeloproliferative disease. Br J Hematol 2007;139:350.
19. Cervantes F. Modern management of myelofibrosis. Br J Haematol 2004;128:583-92.
20. Hasselbalch HC, Clausen NT, Jensen BA. Successful treatment of anemia in idiopathic myelofibrosis with recomibinant human erythropoietin. Am J Hematol 2002;70:92-9.
21. Mesa RA, Steensma DP, Pardanani A, Li CY, Elliot M, Kaufmann SH, Wiseman G, Gray LA, Schroeder G, Reeder T, Zeldis JB, Tefferi A. A phase 2 trial of combination low-dose thalidomide and prednisolone for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003;101:2534-41.
22. Mesa RA, Nagorney DS, Schwager S, Allred J. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer 2006 Jul 15;107(2):361-70.
23. Merup M, Lazarevic V, Nahi H, Andreasson B, Malm C, Nilsson L, Brune M, LeBlanc K, Kutti J, Birgegård G; Swedish Group for Myeloproliferative Disorders. Different outcome of allogeneic transplantation in myelofibrosis using conventional or reduced-intensity conditioning regimens. Br J Haematol 2006; 135; 367-73.
24. Siragusa S, Passamonti F, Cervantes F, Tefferi A. Survival in young patients with intermediate-/high-risk myelofibrosis: Estimates derived from databases for non transplant patients. Am J Hematol 2008;84:140-143.
25. Rambaldi A, Barbaui T, Barosi G. From palliation to epigenetic therapy in myelofibrosis. Amerian Society of Hemtology Education Program Book 2008.
26. Wang J-C, Chen C, Dumlao T el al. Enhanced HDAC enzyme activities in blood CD34+ cells in patients with myeloid metaplasia myelofibrosis. Blood 2007; 110. Abstract #4657.
26. Merup M, Löfvenberg E, Palmblad J: Nya rekommendationer om kroniska myeloproliferativa sjukdomar – Ny kunskap måste ge ny praxis. Läkartidn 2009:106:98-9.
27. Le Bousse-Kerdilès MC, Martyré MC, Samson M Cellular and molecular mechanisms underlying bone marrow and liver fibrosis: a review. Eur Cytokine Netw. 2008;19:69-80
367.