Sammanfattat
Kliniska studier delas ofta in i deskriptiva studier, som används för att formulera en klinisk/biologisk hypotes, och analytiska studier (randomiserade eller observationella) vars primära syfte är att testa en hypotes.
För läkemedelssäkerhet finns ingen studiedesign som är »bättre« eller »sämre« än någon annan. Valet av studiedesign beror på frågeställningen.
Målsättningen för kliniska studier är att kombinera hög intern validitet med hög extern validitet. Intern validitet avser att studien inte har systematiska fel som leder till över- eller underskattning av sambandet mellan exponering och utfall, att inga faktorer stör sambandet och att precisionen är god. Extern validitet avser att det samband som uppvisas gäller för de patientgrupper man önskar applicera resultaten på.
Varje studie måste bedömas utifrån sina individuella styrkor och svagheter. Detta kräver ämnes- och metodkunskap.
Studier av läkemedelssäkerhet utgör en provkarta över olika studiedesigner. Här lånar vi exempel från området läkemedelssäkerhet för att illustrera en del av de metodologiska utmaningar som valet av studiedesign erbjuder. Fokus ligger på möjligheter och erfarenheter av att studera läkemedelssäkerhet med hjälp av observationella kohortstudier, men även styrkor och svagheter med andra typer av studiedesign diskuteras.
Studiedesignens taxonomi. Förenklat kan kliniska studier delas in utifrån design (Figur 1). En deskriptiv studie är beskrivande. Den avser inte att testa en biologisk/klinisk hypotes, men kan vara instrumentell för att formulera en sådan. Fallrapporter, prevalens- eller incidensstudier är exempel på deskriptiva studier. Analytiska studier avser att mäta ett hypotetiserat orsakssamband mellan en exponering (x) och ett utfall (y), det vill säga att testa en hypotes. De kan i sin tur delas in i interventionsstudier, där exponeringen aktivt fördelas bland studiepersonerna, och i observationella studier, där fördelningen av exponering inte styrs av studieledarna.
En randomiserad klinisk prövning är ett typexempel på en interventionsstudie, medan kohortstudier och fall–kontrollstudier är exempel på observationella studier. En randomiserad klinisk prövning kan dock ses som en typ av kohortstudie där exponeringen fördelats på ett kontrollerat men slumpmässigt sätt. Studien följer en grupp individer med olika exponeringar över tid med avseende på uppkomst av ett utfall.
För vissa analytiska studier är valet mellan intervention eller observation självklart. Det är exempelvis svårt att aktivt fördela något som redan är givet, till exempel genotyp, men lättare att observera effekten (med avseende på utfallet) av en given genotyp såsom den fördelats i befolkningen. Även om det i andra sammanhang går att göra en randomiserad interventionsstudie sätter ibland logistik, etik eller kostnader stopp. Då blir observationell studiedesign det enda rimliga alternativet.
Studiedesign och hierarki. Arbete med läkemedelssäkerhet illustrerar att man bör se på olika studiedesigner som mer eller mindre ändamålsenliga och komplementära beroende på frågeställningen. I den ideala analytiska studien motsvarar det uppmätta sambandet mellan exponering och utfall ett orsakssamband; studien uppvisar intern validitet. I verkligheten måste man alltid fråga sig om avsaknad av samband kan förklaras av bristande statistisk precision (vilket oftast syns i form av breda konfidensintervall och höga p-värden) eller någon form av systematiskt fel (bias) som leder till över- eller underskattning av sambandet mellan x och y oavsett hur stor man gör studien.
Förväxlingsfaktorer – att exponeringen är förknippad med en annan faktor, som i sig var en störfaktor för utfallet och därför stör mätningen av sambandet mellan exponering och utfall – är kanske det mesta kända exemplet. Bortom systematiska fel och störfaktorer måste man för ett uppmätt samband som man på goda grunder uppfattar som ett orsakssamband fråga sig för vilka patientgrupper detta samband gäller. Dessa frågor rör studiens generaliserbarhet, eller dess externa validitet.
Hotbilden mot den interna och externa validiteten är dock lite olika för olika studiedesigner, men viktigt är att varje studie måste bedömas utifrån sina individuella styrkor och svagheter. Studier av läkemedelssäkerhet kräver därför både ämneskunskap (vad är ändamålsenligt givet den biologiska eller kliniska frågeställningen?) och metodkunskap (hur ska studien utformas och vad har studiedesignen för styrkor och svagheter?).
Vad krävs för att studera läkemedelssäkerhet?
Sannolikheten att identifiera en biverkan bestäms av faktorer som tidssamband, hur allvarlig/iögonfallande en biverkan är, hur vanligt samma tillstånd är hos obehandlade individer och hur noga man letar, men också av vad man betraktar som en biverkan. Många biverkningar som i Fass-texten listas som vanliga uppkommer tidigt, med ett tydligt tidssamband till läkemedelsintaget. Dessa kan därför lätt identifieras och kvantifieras redan i det kliniska prövningsprogram som föregår läkemedlets godkännande. Andra biverkningar är helt oväntade, mindre vanliga och/eller uppträder efter lång tid och är därför kanske inte kända vid tidpunkten för läkemedlets godkännande. Spontanrapportering av biverkningar och fallrapporter från uppmärksamma kliniker är de viktigaste informationskällorna för upptäckt av allvarligare former av sådana biverkningar. Ett exempel är signalen om narkolepsi efter vaccination med Pandemrix i samband med influensapandemin 2009, något som identifierades via spontanrapporter [1].
En biverkan måste inte vara kopplad till läkemedlet som sådant utan kan uppkomma exempelvis genom att det uppstår ett fel i tillverkningen av substansen eller något hjälpämne. Vid så kallad signalspaning kan detta identifieras genom ett plötsligt ökat antal rapporterade fall av en specifik biverkan hos ett sedan länge etablerat läkemedel. I de stora databaserna över spontanrapporterade biverkningar, EudraVigilance [2] och det svenska biverkningsregistret Swedis [3], sker kontinuerligt sådan signalspaning.
För att en biverkan i klinisk praxis ska identifieras och rapporteras av patienten/doktorn krävs att den som rapporterar gjort en koppling mellan behandlingen i fråga och det tillstånd som uppstått. Om tiden mellan insättning av läkemedlet och biverkans uppträdande (latenstiden) är lång är det möjligt att den som identifierar symtomet inte är varse den behandling som initierade biverkan. Biverkningsbilden kan förekomma i relativt hög prevalens även hos den obehandlade delen av befolkningen. Tillsammans med ett svagt tidssamband och polyfarmaci gör det kopplingen till ett specifikt läkemedel svår att upptäcka. Förutsättningarna för klassisk läkemedelsuppföljning via biverkningsrapportering blir då begränsade. Ett viktigt exempel är upptäckten att åtminstone vissa NSAID tycks vara associerade med kardiovaskulära biverkningar [4, 5].
Det har blivit alltmer tydligt att vi också måste vidga begreppet biverkan bortom de tillstånd som direkt och enbart kan tillskrivas ett läkemedel. Det kan till och med vara svårt att hålla isär effekt och bieffekt av behandling, i synnerhet vid kroniska sjukdomar där mycket av morbiditeten utgörs av följd- och samsjuklighet. Patienter med reumatoid artrit löper en ökad risk att utveckla infektioner, dels på grund av faktorer som har med sjukdomen och dess aktivitet att göra, dels på grund av dess behandling. Ett immundämpande läkemedel som påtagligt förbättrar sjukdomskontrollen och därmed sänker risken för infektion kan samtidigt i sig, via den immundämpande effekten, öka risken för infektion. Nettoeffekten med avseende på infektionsrisk, det vill säga den observerade förekomsten av infektioner hos behandlade patienter, kan vara såväl positiv som negativ. Den kan dessutom variera mellan olika patientgrupper och över tid sedan behandlingsstart.
Läkemedelssäkerhet blir i denna bemärkelse intimt förknippad med läkemedelseffekt och samsjuklighet, och biverkan är inte längre ett tillstånd hos en enskild patient utan måste förstås på gruppnivå. Behandlingen i exemplet ovan leder till att vissa som skulle ha utvecklat en infektion inte gör det, medan andra som annars inte skulle utvecklat en infektion gör så. Risk–nyttakvoten kan alltså variera mellan olika patientgrupper med samma grundsjukdom. Observationella studier, och tillgång till kliniska data kring behandling, effekt och följdsjuklighet över lång tid, blir i detta perspektiv ett helt nödvändigt komplement till kliniska prövningar, fallrapporter och spontanrapportering av klassiska biverkningar.
Var studiedesign på sin plats
Deskriptiva studier som fallrapporter och spontanrapportering utgör ett viktigt sätt att identifiera möjliga biverkningar, även om begränsningarna är flera. Utan tillgång till information om den samlade exponeringen för läkemedlet blir det svårt att skatta risken per antal behandlade och utan information om förekomst av samma tillstånd hos obehandlade blir det svårt att skatta styrkan i ett samband, det vill säga hur mycket förhöjd risken är jämfört med individer som inte är exponerade. Den viktigaste begränsningen är dock kanske underrapportering; dels av biverkningar som kanske anades, men inte rapporterades, dels av tillstånd som delvis utlösts av behandlingen men som inte uppfattats som en biverkan på individnivå. Ett sådant exempel är okomplicerad pneumoni hos en multisjuk och immunsupprimerad patient med reumatoid artrit.
Experimentella studier. Bland de analytiska studierna har den randomiserade kliniska prövningen uppenbara fördelar. En lyckad randomisering i en tillräckligt stor studie gör de grupper som jämförs lika i alla andra avseenden än exponeringen. En skillnad i utfall mellan dessa grupper bör därför kunna tillskrivas just exponeringen. Randomiserade kliniska prövningar är utmärkta medel att såväl identifiera som kvantifiera biverkningar med kort latens och hög förekomst.
I många fall är dock även en stor klinisk prövning både för liten och för kortvarig för att kunna skatta läkemedelssäkerhet (Figur 2). Detta gäller i synnerhet för läkemedel avsedda för behandling av kroniska sjukdomar där en stor och lång klinisk prövning kan bestå av några hundra patienter med upp till ett års uppföljning, men där tusentals patienter i klinisk praxis inte sällan behandlas i fem, tio år eller mer.
Inom flera terapiområden uppstår frågetecken kring generaliserbarheten av kliniska prövningar. Studiepopulationerna är ofta kraftigt selekterade och inkluderar individer som har en bättre/sämre prognos eller är på annat sätt annorlunda, exempelvis avseende andra riskfaktorer och samsjuklighet, än den tilltänkta behandlingspopulationen. Inte sällan görs kliniska prövningar i länder med annan riskfaktorprofil än den hos svenska patienter. Det kan vara värt att komma ihåg att en randomiserad studie är balanserad med jämförbara »armar« vid studiestarten, men inte nödvändigtvis därefter. Om till exempel andelen patienter som avbryter studien över tid skiljer sig mellan behandlingsarmarna, kommer validiteten att hotas enligt samma principer som i en observationell kohortstudie.
»Long-term extension studies«, studier där man fortsatt att följa patienter som ingått i en klinisk prövning även efter prövningens slut, är ett sätt att komma runt den korta uppföljningstiden i prövningen. Denna typ av studie är av värde för att studera exempelvis varaktigheten i respons, men erbjuder endast begränsat med information vad gäller säkerhet. Oftast följs endast behandlingsarmen (det finns ingen jämförelsegrupp), och hela studien är betingad av selektionen in i den ursprungliga kliniska prövningen, som skärpts ytterligare genom att endast de som är kvar i prövningen vid dess slut (patienter med respons men inga biverkningar) går in i uppföljningsfasen.
Metaanalyser av flera välgjorda randomiserade kliniska studier ökar studiepopulationens storlek och kan besvara frågan om mer ovanliga biverkningar. Det är viktigt att komma ihåg att patienterna är lika selekterade som i de kliniska prövningar som ingår och att uppföljningstiden är oförändrad. Metaanalyser kan därför inte ge oss ledtrådar avseende risk i det långa loppet och kanske inte för risker hos patienter i klinisk praxis.
Observationella studier. Observationella studier, i synnerhet om de baserar sig på redan existerande datakällor som till exempel register, möjliggör större studiepopulationer och längre uppföljningstider och därmed förbättrade möjligheter att identifiera och kvantifiera ovanliga men allvarliga biverkningar även efter lång tid (Figur 2). De ger också möjligheter att studera mindre selekterade patientpopulationer, liksom det omvända; att specifikt identifiera patientgrupper med vissa karaktäristika som kanske exkluderats från kliniska prövningar. Framför allt ger observationella studier möjlighet att studera läkemedelssäkerhet i den lite vidare bemärkelsen i termer av ändrade mönster av samsjuklighet.
En utmaning och potentiellt problem vid läkemedelssäkerhetsanalyser baserade på observationella data är så kallad confounding by indication, vilket innebär att indikationen för behandling är associerad med utfallet. Vad det är som orsakar utfallet (indikationen eller behandlingen) blir i dessa fall svårt att urskilja. Eftersom vi i klinisk praxis inte lottar patienter till olika behandlingsstrategier utan triagerar dem efter medicinskt behov, är de som behandlas ofta sjukare än de som inte behandlas. Att då jämföra behandlade patienter med obehandlade blir som att jämföra äpplen med päron. Även med avancerade statistiska tekniker kan det då vara svårt att förstå om ett orsakssamband föreligger mellan behandling och utfall. Valet av jämförelsegrupp är därför den största utmaningen för observationella studier. I många fall varierar risken för biverkan/följdsjuklighet över tid. Det är därför viktigt att studera risken som en funktion av tid sedan behandlingsstart snarare än som den genomsnittliga risken totalt sett. En inte ovanlig felkälla vid analys av läkemedelssäkerhet i observationella studier är »left truncation«, det vill säga att man följer patienter som är under (snarare än startar) behandling vid en given tidpunkt med avseende på ett säkerhetsutfall [6]. Risken är då stor att den behandlade population som följs är »urtvättad« med avseende på tidiga biverkningar och riskökningar.
Även av andra skäl är tiden kring behandlingsstart ofta speciell. Till exempel förutsätter många behandlingar först en kontroll av några blodprov eller kanske en lungröntgen, och kanske kommer patienten på tätare återbesök. Detta leder till högre sannolikhet att upptäcka en begynnande biverkan, till exempel lungcancer, redan före behandlingen och då exkluderas ur studien. Följden blir att de som faktiskt startar behandling kommer att ha en låg »urtvättad« korttidsrisk för just lungcancer. Det blir också svårt att upptäcka en biverkan tidigare än vad som annars skulle varit fallet efter behandlingsstart. Om möjligt bör man därför alltid försöka följa den behandlade gruppen från behandlingsstart, och göra på exakt samma vis med jämförelsegruppen som kanske valts ut på basen av att de startat en alternativ behandling. Observationella studier möjliggör alltså stora och populationsbaserade studier över lång tid och därmed möjligheter att identifiera och kvantifiera ovanliga biverkningar, liksom att skatta förändringar i samsjuklighet i relation till läkemedelsbehandling. Okritisk analys av observationella data medför stor risk att felaktiga slutsatser dras. Det är alltså viktigt att bedöma varje individuell studie utifrån de metodologiska utmaningar som följer av problemställningen och hur dessa tacklats. Förekomst av störfaktorer eller andra systematiska fel är i sig inte ett skäl att avfärda en studie; det är viktigare att förstå hur de kan ha påverkat resultatet.
Observationella kohortstudier i Sverige
Sverige erbjuder goda möjligheter att genomföra högkvalitativa studier av läkemedelssäkerhet på stora patientgrupper som följs över lång tid. Socialstyrelsen har i decennier registrerat hälsodata som födslar, cancerfall, dödsfall och slutenvård. På senare tid har man även skapat rikstäckande register över förskrivningsläkemedel och öppenvård (exklusive primärvård). Dessutom finns för många sjukdomar professionsdrivna kvalitetsregister med detaljerade sjukdoms- och behandlingsspecifika data. Kvalitetsregister kan erbjuda detaljerade sjukdomsspecifika mått på exempelvis sjukdomens svårighetsgrad och aktivitet som de nationella hälsodataregistren inte har.
Med personnumret kan datakällor länkas samman för att monitorera läkemedelssäkerhet för patienter som får behandling i klinisk praxis. Säkerhetsutfall som exempelvis cancerrisk kan följas upp över lång tid för patienter i Sverige som behandlas med ett visst läkemedel. Ett exempel är uppföljningen av TNF-hämmare, vilka används vid behandling av bland annat reumatiska sjukdomar [7]. Randomiserade kliniska prövningar av dessa högpotenta immunmodulerande läkemedel inkluderade oftast bara några hundra patienter, varade sällan längre än ett år och inkluderade initialt de absolut svårast sjuka patienterna (utan alltför komplicerande samsjuklighet), varför inte ens stora metaanalyser konklusivt kunnat besvara frågan om vad som händer efter första årets behandling [8].
Med hjälp av Svensk reumatologis kvalitetsregister har hittills närmare 20 000 patienter kunnat följas avseende bland annat cancerrisk [9]. Registret inkluderar såväl nyinsjuknade i övrigt relativt friska patienter i fertil ålder som långtidssjuka patienter över 90 år med omfattande samsjuklighet. Cancerutfall kan observeras via Socialstyrelsens cancerregister under lång tid, med mycket litet bortfall (»loss to follow-up«). Matchade jämförelsegrupper kan identifieras med en kombination av data från Socialstyrelsens och Statistiska centralbyråns register. Därigenom kan många potentiella störfaktorer elimineras och mer jämförbara grupper genereras.
Observationella kohortstudier är ett viktigt komplement till de säkerhetsdata som kan erhållas från randomiserade prövningar och spontanrapportering, men de ställer analytiska utmaningar. I Sverige har den kliniska professionen i samarbete med epidemiologisk kompetens stora möjligheter att lämna avgörande bidrag till vår förståelse av effekt och bieffekt av behandling i klinisk praxis. I tider av viss dysterhet kring läget för svensk klinisk forskning är denna nisch ett ljus i mörkret.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
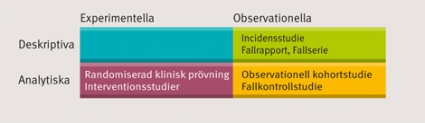
Figur 1. Schematisk uppdelning av de vanligast förekommande typerna av studiedesign.
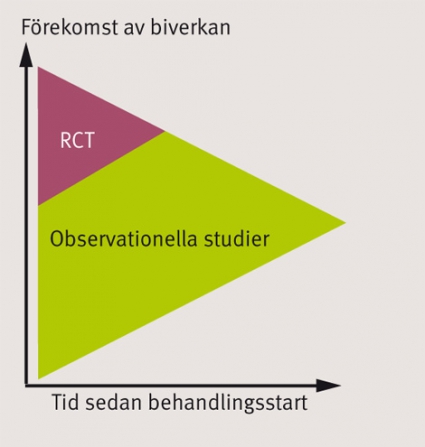
Figur 2. I randomiserade kliniska prövningar studeras vanligen mer vanliga biverkningar (y) under en kortare tid (x). Observationella kohortstudier kan göras större med längre uppföljningstid. För bägge gäller att antalet individer som nått en given uppföljningstidpunkt tenderar att minska över tid. Därmed avtar förmågan att skatta (ovanliga) risker.
(uppdaterad 2021-09-01)
Referenser
1. Bardage CI, Persson A, Örtqvist A, et al. Neurological and autoimmune disorders after vaccination against pandemic influenza A (H1N1) with a monovalent adjuvanted vaccine: population based cohort study in Stockholm, Sweden. BMJ. 2011;343:d5956.
2. EudraVigilance. Pharmacovigilance in the European Economic Area. London: European Medicines Agency; 2012. http://eudravigilance.ema.europa.eu/highres.htm
3. Data mining som redskap för signalgenerering. Uppsala: Läkemedelsverket; 2017. http://www.lakemedelsverket.se/Alla-nyheter/NYHETER-2007/Data-mining-som-redskap-for-signalgenerering/
4. Jüni P, Nartey L, Reichenbach S, et al. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364(9450):2021-9.
5. Fosbøl EL, Køber L, Torp-Pedersen C, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs among healthy individuals. Expert Opin Drug Saf. 2010;9(6):893-903.
6. Ray WA. Evaluating medication effects outside of clinical trials: new-user designs. Am J Epidemiol. 2003;158(9):915-20.
7. Askling J, van Vollenhoven RF, Granath F, et al. Cancer risk in patients with rheumatoid arthritis treated with anti-tumor necrosis factor alpha therapies: does the risk change with the time since start of treatment? Arthritis Rheum. 2009;60(11):3180-9.
8. Askling J, Fahrbach K, Nordström B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20(2):119-30.
9. Neovius M, Simard J, Sundström A, et al. Generalisability of clinical registers used for drug safety and comparative effectiveness research: coverage of the Swedish Biologics Register. Ann Rheum Dis. 2011;70(3):516-9.
Summary
Clinical studies are often categorized as either descriptive, instrumental for generating hypotheses, or analytic, aiming to test such hypotheses. When working with treatment-safety there are no study designs that are always ”better” or ”lesser” suited than others. The choice of study design should be based on the research question at hand.
The aim of any clinical study, regardless of design, is that they have a high internal validity (void of systematic errors leading to over- or underestimations of the true association between exposure and outcome, that there are no confounders of the true association, and that the precision is good) and external validity (the association is true also for the groups of patients in whom the results are meant to be applied).
Threats to internal and external validity differ by study design. Every study – regardless of design –must always be criticized based on its individual strengths and weaknesses. To do this, both subject matter and methodological knowledge are needed.
Marie Holmqvist, Pauline Raaschou, Martin Neovius, Johan Askling
Correspondence: Johan Askling, Enheten för klinisk epidemiologi, Karolinska universitetssjukhuset, Solna, SE-171 76 Stockholm, Sweden
johan.askling@ki.se