Vid subakut till akut konfusion med fokala epileptiska anfall – överväg anti-LGI-1-encefalit.
Diagnosen ställs genom påvisande av neuronala autoantikroppar i serum och/eller likvor.
Anti-LGI-1-encefalit manifestar sig ofta med fokala brakiofaciala motoriska epileptiska anfall samt dysautonoma symtom såsom pilomotoriska anfall med rysningar och gåshud.
Förändringar förenliga med anti-LGI-1-encefalit ses ofta (56–90 procent) i mediala delar av temporalloberna på MR–hjärna (T2/FLAIR och diffusionsviktad sekvens).
EEG är oftast patologiskt.
Immunterapi bör påbörjas snarast.
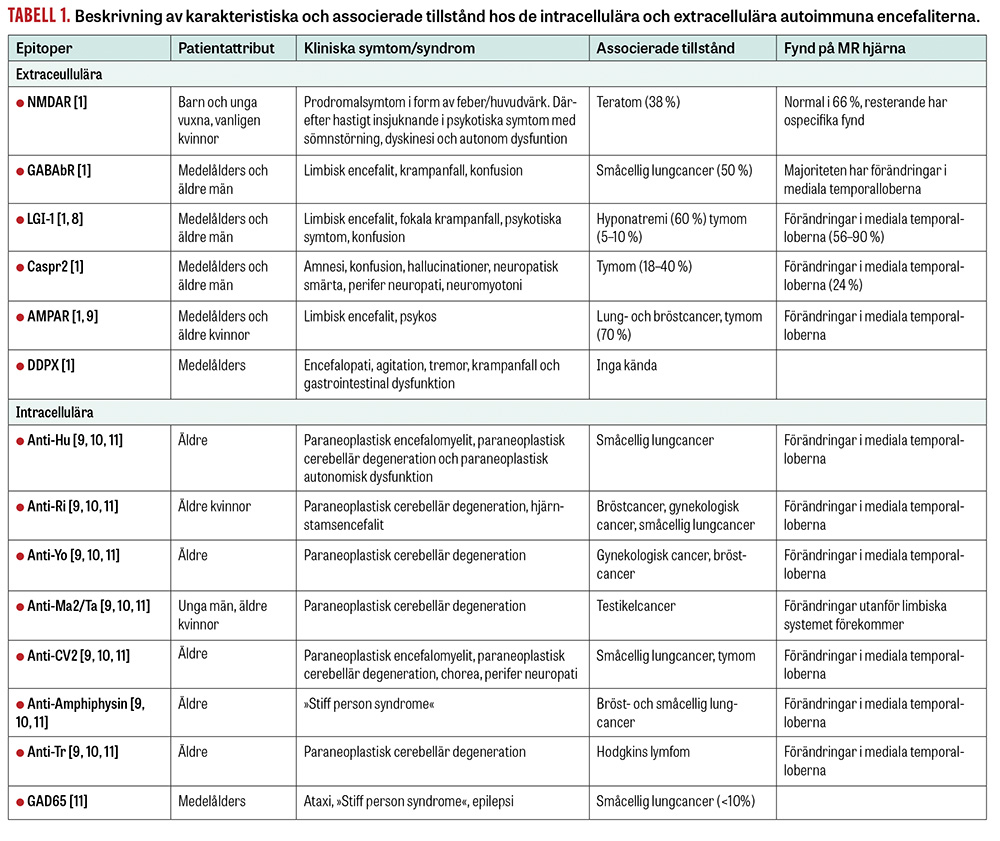
Introduktion. Autoimmun encefalit är ett samlingsnamn för inflammation i hjärnan orsakat av antikroppar mot epitoper på protein i synapser eller på neuronens cellyta [1,2]. Hittills identifierade epitoper är de extracellulära NMDAR (N-metyl-D-aspartatreceptor), GABAbR (gamma-aminosmörsyra-B-receptor), LGI-1 (leucinrikt gliominaktiverat protein-1), CASPR2 (kontaktinassocierat protein 2), AMPAR (alfa-amino-3-hydroxi-5-metyl-4-isoxazolpropionsyrareceptor) och DDPX (dipeptidylpeptidaslikt protein 6) samt de intracellulära paraneoplastiska anti-Hu, anti-Ri, anti-Yo, anti-MA2/Ta, anti-CV2, anti-amfifysin, anti-Tr och GAD65 (65 kDa-isotypen av glutaminsyradekarboxylas). Karakteristika för de olika autoimmuna encefaliterna går att läsa om i Tabell 1. Vid autoimmuna encefaliter är det limbiska systemet ofta påverkat med symtom som konfusion och psykos, vilket har gett upphov till beteckningen limbisk encefalit. Ett exempel är anti-LGI-1-antikroppsmedierad encefalit, där antikropparna riktas mot ett cellyteprotein i komplexet av spänningskänsliga kaliumkanaler, där LGI-1 är det vanligaste proteinet [2].
Insjuknandet i anti-LGI-1-encefalit är subakut till akut med symtom i form av minnessvikt, förändringar i beteende i form av psykosliknande tillstånd, konfusion samt epileptiska anfall [1-4]. Ungefär 10 procent av patienter med antikroppar mot LGI-1 har en underliggande tumörsjukdom, och tymom är vanligast [5-7]. Det är vanligt med samtidig behandlingsrefraktär hyponatremi i form av SIADH (syndrome of inapprop-riate sectretion of antidiuretic hormone) [2]. Diagnosen anti-LGI-1-encefalit ställs genom påvisande av antikroppar mot LGI-1 i serum eller cerebrospinalvätska [1]. Flertalet studier rapporterar om att tecken till anti-LGI-1-encefalit ofta (56–90 procent) ses på magnetkameraundersökning (MR) av hjärnan, som uttryck för en inflammatorisk process i limbiska systemet. Signalförändringar i temporalloben vid anti-LGI-1-encefalit ses vid MR-undersökning av hjärna med T2/FLAIR och diffusionsviktad sekvens [7, 8, 12].
Nedan presenteras ett fall med en 68-årig man som uppvisade symtom i form av attacker av gåshud och konfusion. Syftet med denna fallbeskrivning är att öka uppmärksamheten på att anti-LGI-1-encefalit kan vara orsaken till konfusion och fokala epileptiska anfall.
Fallbeskrivning
I juni 2018 sökte en 68-årig man akutmottagningen på Hässleholms sjukhus på grund av konfusion, frossa och feber. Sedan tre månader hade patienten haft anfall i form av gåshud, frossa, mörkräddhet och ångest. Anfallen av gåshud kom utan förvarning upp till tio gånger dagligen, varade i någon enstaka minut och uppträdde framför allt i armar och huvud. Under anfallen blev patienten också mycket orolig och ångestfylld. Patienten uppvisade även paranoida tankar, vilket gjorde att han sökte både kirurg- och medicinakutmottagning upprepade gånger under våren 2018. Till slut remitterades han vidare till psykiatrisk akutmottagning på grund av sin oro och mörkräddhet, där han ordinerades bensodiazepiner som inte hade någon effekt alls.
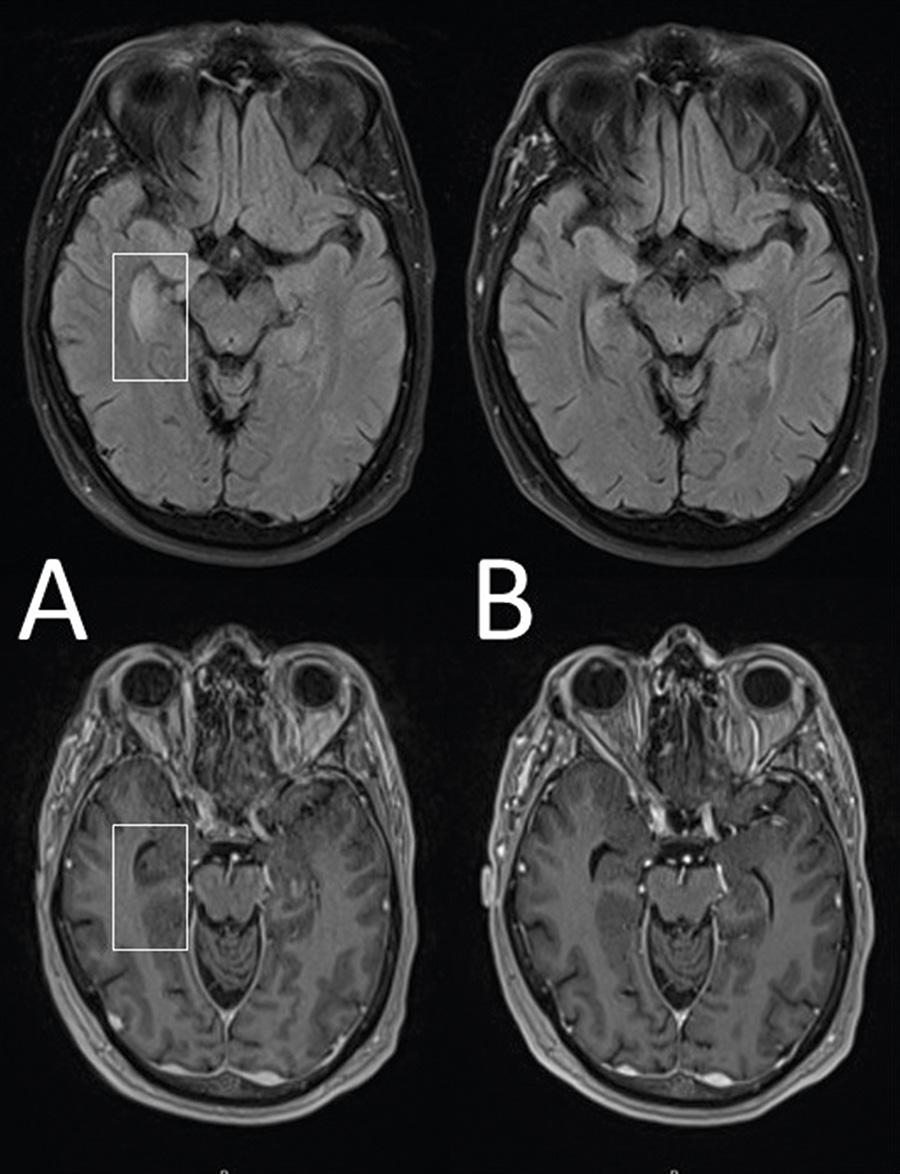
Patienten sökte akutmottagningen i juni 2018 på grund av konfusion, frossa och feber och lades då in på medicinavdelning i Hässleholm för utredning. Patienten var desorienterad till tid, rum och situation men ej till person. Han rapporterade upp till 10 stycken minutlånga attacker av gåshud och ångest dagligen. Rutinnervstatus och basprov var normala frånsett stegrat glukos. Likvor var utan hållpunkter för bakteriell eller viral meningit, och cellräkning, laktat- och glukoshalt var normala. PCR var negativ för herpes simplex virus 1 och 2 samt varicella–zoster-virus. Serologi för borrelia och TBE var negativa. MR-hjärna visade högsignalerande förändringar i höger temporallob (se Figur 1, A) som väckte misstanke om autoimmun encefalit. Kompletterande blodprov för neuronala autoantikroppar påvisade LGI-1-antikroppar i serum (titer 1/100). Diagnosen anti-LGI-1-encefalit ställdes, och behandling med intravenöst immunglobulin (IVIG) (Privigen 0,4g/kg i 3 dagar var 4:e vecka) påbörjades. Datortomografi av torax och buk var utan tecken till tumör. Patienten förbättrades kognitivt efter behandlingen och skrevs ut med fortsatt uppföljning via neurologimottagningen.
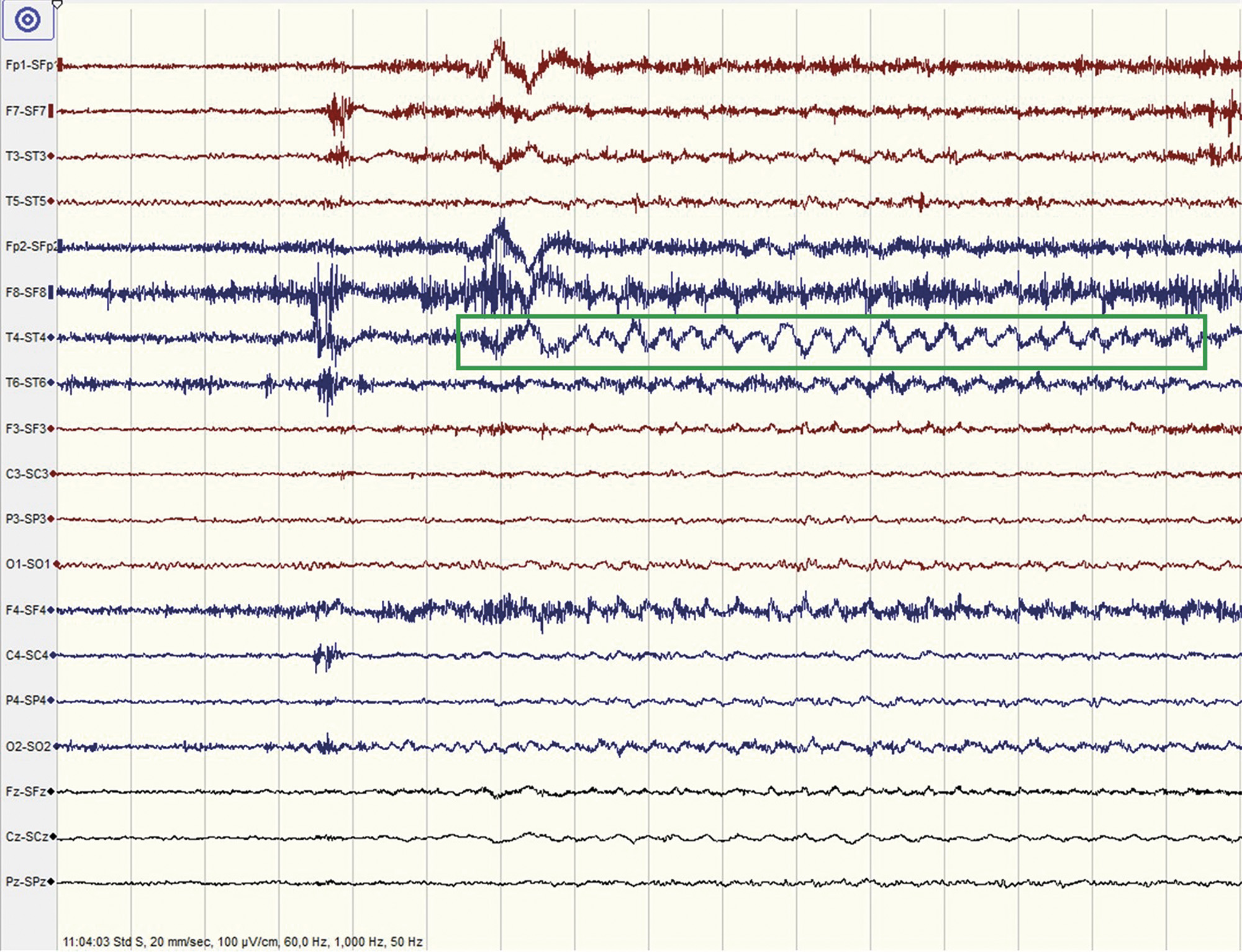
Under hösten 2018 ringde patienten flertalet gånger till neurologimottagning på grund av återkommande attacker av gåshud och kvarstående lättare kognitiv dysfunktion. Då anti-LGI-1-antikroppar fortfarande kunde påvisas i serum (titer 1/100) och patienten inte var symtomfri avslutades IVIG, och i stället påbörjades behandling med metylprednisolon intravenöst (1 g var fjärde vecka). Patienten upplevde initialt en minskning av sina anfall, men hörde efter kort tid av sig på grund av försämring och otillräcklig symtomkontroll. I november, i samband med återbesök hos neurolog, tog han åter upp attackerna av gåshud och beskrev dessa som kraftiga rysningar som började i en arm som därefter med kort fördröjning spred sig till bål, huvud och andra armen. Han berättade att dessa varade ungefär en minut och återkom upp till 15-20 gånger dagligen. Attackerna åtföljdes av kraftig oro och hjärtklappning. I samband med besöket fick patienten en sådan typisk attack som väckte misstanke om epileptiskt anfall av pilomotorisk typ. Mot bakgrund av epilepsimisstanken utfördes EEG, som visade en 20 sekunder lång episod med elektrografisk anfallsaktivitet med dominans temporalt höger sida som startade ca 10 sekunder efter symtomdebut i form av rysningar i kroppen och gåshud (se Figur 2). Sammantaget tolkades således episoderna av gåshud som ett epileptiskt anfallsuttryck. Därför satte man in levetiracetam under fortsatt behandling med metylprednisolon. Förnyad kontroll av antikroppar mot LGI-1 visade oförändrad titer på 1/100, och då patientens pilomotoriska anfall återkom i oförändrad omfattning togs beslut om att avsluta behandlingen med metylprednisolon. Då den patofysiologiska mekanismen sannolikt var antikroppsmedierad beslutades om byte till behandling med rituximab, som gav upphov till en snabb och kraftfull B-cellssuppression. Denna behandling startades i januari 2019, och efter tre månader sågs sjunkande nivåer av anti-LGI-1-antikroppar (titer 1/10). På grund av biverkningar skedde under våren byte från levetiracetam till karbamazepin följt av valproat. Då inte heller detta tolererades skedde i maj byte till lamotrigin som antiepileptisk behandling, vilket patienten inte upplevt biverkningar av. Inga ytterligare anfallsmanifestationer har noterats sedan dess.
Uppföljande MR i juni 2019 visade regress av tidigare beskrivna förändringar i temporalloben (se Figur 1, B). PET-DT utförd ungefär samtidigt var utan tecken till malignitet. Efter påbörjad behandling med rituximab förbättrades patienten kliniskt och de fokala anfallen av gåshud och ångest minskade, vilket åtföljdes av sjunkande titer av anti-LGI-1-antikroppar.
Diskussion
Autoimmun encefalit beskrevs första gången på 1960-talet [13]. Nyligen föreslogs reviderade diagnoskriterier [5]. De första tre kriterierna nedan föreslås vara nödvändiga för diagnosen anti-LGI-1 encefalit:
- ett subakut insjuknande (mindre än tre månader) i kognitiv dysfunktion, epileptiska anfall och psykiatriska symtom,
- bilaterala förändringar medialt i temporalloben på MR-hjärna,
- pleocytos i cerebrospinalvätskan och/eller EEG-mönster talande för epileptisk eller långsam aktivitet temporalt, och
- andra diagnoser som herpesencefalit, Whipples eller Behçets sjukdom har uteslutits.
Diagnosen limbisk encefalit kan även ställas, utan att ovanstående kriterier är uppfyllda, genom att antikroppar mot synaptiska eller cellyteprotein påvisas i serum eller likvor. Vid välgrundad klinisk misstanke utan fynd av antikroppar och icke uppfyllda diagnoskriterier kan ändå empirisk behandling påbörjas. Differentialdiagnoser till limbisk anti-LGI-1-encefalit är, förutom de beskrivna autoimmuna encefaliterna i tabell 1, även Hashimotos encefalopati, Creutzfeldt–Jakobs sjukdom, Susacs syndrom, Wernicke–Korsakoffs syndrom, CNS-vaskulit, CNS-lymfom och CNS-infektion [1].
Säkra incidens- och prevalensuppgifter saknas för autoimmuna encefaliter, då det sannolikt finns en betydande underdiagnostik. Beräknad incidens är 5–10 per 100 000 invånare och år [1]. Dubbelt så många män som kvinnor insjuknar i anti-LGI-1-encefalit, oftast runt 60 års ålder, enligt publicerade fallserier [7, 12]. Framträdande symtom vid anti-LGI-1-encefalit är kognitiv dysfunktion med minnessvikt och psykiska symtom liknande psykos, pilomotorisk epilepsi, och brakiofaciala dystona anfall. Utöver de neuropsykiatriska manifestationerna förekommer allvarlig hyponatremi i 60–88 procent av fallen [1, 5, 6]. Patienten i fallbeskrivningen ovan hade ingen hyponatremi men däremot psykiatriska symtom i form av paranoida tankar och även viss minnespåverkan. De flesta (80 procent) med anti-LGI-1-encefalit uppvisar pleocytos i likvor, och drygt hälften har oligoklonala band. Likvoranalys kan dock vara helt normal. [14].
Vid anti-LGI-1-encefalit är EEG oftast patologiskt med antingen fokal epileptiform eller förlångsammad aktivitet relaterad till ett eller flera epileptiska foci [2]. Patientens anfallssemiologi innefattade attacker av gåshud, så kallad pilomotorepilepsi. Flera studier har beskrivit att epileptiska anfall ledande till aktivering av pilomotormuskeln är vanligt förekommande vid epileptisk aktivitet i temporalloberna [3, 15, 16]. Om pilomotoriska anfall är dominerande anfallssemiologi är detta närmast patognomont för anti-LGI-1-encefalit [15]. Det är okänt hur LGI-1-antikroppar orsakar epilepsi [3], men det är känt att mutationer i genen för LGI-1 orsakar autosomalt dominant lateral temporallobsepilepsi [4,7].
Patienten i fallbeskrivningen ovan blev först behandlad med IVIG, men på grund av utebliven effekt skedde byte till steroider och slutligen rituximab. Snabb diagnos och insättande av intensiv immunterapi kan både bota anti-LGI-1-encefalit och minska risken för kvarstående funktionsnedsättning [5, 17].
Sammanfattningsvis är anti-LGI-1-encefalit ett livshotande tillstånd som obehandlat med stor sannolikhet leder till långsiktig neurologisk funktionsnedsättning. Sjukdomsförloppet varierar från individ till individ, och evidensbaserade behandlingsriktlinjer saknas. I vilken mån utredning och behandling varierar över landet är okänt och kan bland annat styras av lokal kompetens. Vi ser en möjlig risk för att detta kan leda till ojämlikheter i vården. Nationella riktlinjer för handläggning och initial behandling vid autoimmuna encefaliter hade kunnat underlätta vården för denna patientgrupp och skulle kunna tänkas minimera denna risk. Som ett första steg i ett sådant arbete föreslår vi därför tillsättande av en nationell arbetsgrupp.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
(uppdaterad 2020-05-13)
Referenser
- Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404.
- Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann N Y Acad Sci. 2015;1338:94-114.
- Wennberg R, Maurice C, Carlen PL, et al. Pilomotor seizures marked by infraslow activity and acetazolamide responsiveness. Ann Clin Transl Neurol. 2018;6(1):167-73.
- Montojo MT, Petit-Pedrol M, Graus F, et al. Clinical spectrum and diagnostic value of antibodies against the potassium channel related protein complex. Neurologia. 2015;30(5):295-301.
- Wang M, Cao X, Liu Q, et al. Clinical features of limbic encephalitis with LGI1 antibody. Neuropsychiatr Dis Treat. 2017;13:1589-96.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69(5):892-900.
- Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9(8):776-785.
- Schultze-Amberger J, Pehl D, Stenzel W. LGI-1-positive limbic encephalitis: a clinicopathological study. J Neurol. 2012;259(11):2478-80.
- Dalmau J, Rosenfeld MR. Autoimmune encephalitis update. Neuro Oncol. 2014;16(6):771-8.
- Carvalho F, Massano J, Coelho R. Neuropsychiatric symptoms in autoimmune encephalopathies: a clinican’s guide. International Journal of Clinical Neuroscience and Mental Health. Epub 15 sep 2014. doi: 10.21035/icjnmh.2014.1.11.
- Armangue T, Leypoldt F, Dalmau J. Autoimmune encephalitis as differential diagnosis of infectious encephalitis. Curr Opin Neurol. 2014;27(3):361-8.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010;133(9):2734-48.
- Brierley JB, Corsellis JAN, Hierons R, et al. Subacute encephalitis of later adult life. Mainly affecting the limbic areas. Brain. 1960;83(3):357-68.
- Jarius S, Hoffmann L, Clover L, et al. CSF findings in patients with voltage gated potassium channel antibody associated limbic encephalitis. J Neurol Sci. 2008;268(1–2):74-7.
- Rocamora R, Becerra JL, Fossas P, et al. Pilomotor seizures: an autonomic semiology of limbic encephalitis? Seizure. 2014;23(8):670-3.
- Wieser S, Kelemen A, Barsi P, et al. Pilomotor seizures and status in non-paraneoplastic limbic encephalitis. Epileptic Disord. 2005;7(3):205-11.
- Ariño H, Armangué T, Petit-Pedrol M, et al. Anti-LGI1-associated cognitive impairment: presentation and long-term outcome. Neurol-ogy. 2016;87(8):759-65.
Summary
Anti-LGI-1 encephalitis is a type of autoimmune encephalopathy, where antibodies react against the cell surface protein leucine-rich glioma inactivated protein 1 (LGI-1). It presents with a subacute confusion, changes in behaviour, short-term memory deficits and seizures. A piloerectile semiology is common, which has been described as reflecting insular ictal activity. Patients may have temporal lobe abnormalities on brain MRI and EEG. More than half of the patients with limbic encephalitis associated with anti-LGI1 antibodies have hyponatremia. The diagnosis of anti-LGI-1 encephalitis can be made by the detection of antibodies against LGI-1 in serum and/or cerebrospinal fluid. Prompt diagnosis and treatment are important to avoid long-term disability. This case report describes a man with episodes of goose bumps and mild confusion caused by anti-LGI-1 encephalitis.