




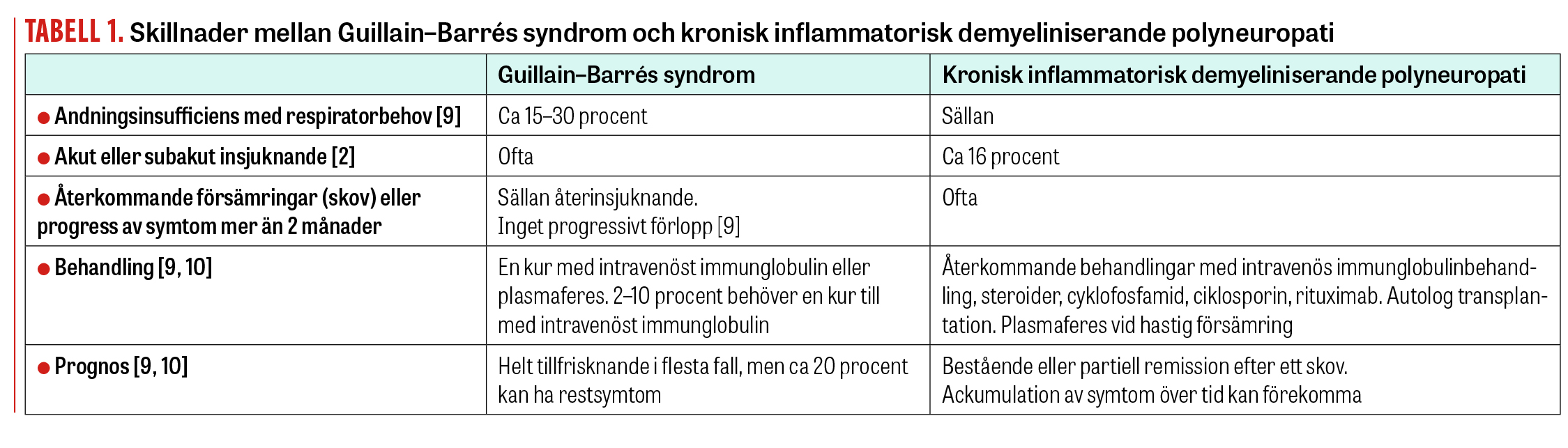
Kontinuerlig uppföljning kan hjälpa oss att skilja Guillain–Barrés syndrom och akut insättande CIDP (A-CIDP), vilket är viktigt med tanke på behandling och prognos (Tabell 1).
CIDP-patienter med tecken till ventilationspåverkan behöver tät övervakning med kontroll av andningsfrekvens, pulsoximetri och PEF (topputandningsflöde), då ventilationsbehandling kan vara indicerad.
Elektroneurografi av frenikusnerv och elektromyografi eller ultraljud av diafragma kan vara till hjälp att identifiera riskpatienter i tid.
Kronisk inflammatorisk demyeliniserande polyneuropati (CIDP) är en autoimmun inflammation av perifera nerver och nervrötter. Den manifesteras som en symmetrisk sensomotorisk polyneuropati med proximal och distal påverkan i samtliga fyra extremiteter och eventuellt även i kranialnerver. Diagnosen ställs med hjälp av anamnes, kliniskt status och elektroneurografi (Fakta 1) [1]. Det finns också en akut form av inflammatorisk polyneuropati som kallas Guillain–Barrés syndrom. Skillnaden mellan CIDP och Guillain–Barrés syndrom är att CIDP förlöper i skov eller har ett mångårigt kroniskt, fortskridande förlopp mer än 2 månader. CIDP och Guillain–Barrés syndrom skiljer sig också vad gäller behandling och prognos (Tabell 1).
Fallbeskrivning
En 56-årig tidigare frisk man sökte akutmottagningen i början av augusti 2020 på grund av stickningar i fötterna sedan 3 dagar tillbaka. Ingen infektion eller trauma i anamnesen. Neurologiskt status och rutinblodprov var normala. Patienten skickades hem med uppföljning på sin vårdcentral.
En vecka senare kom han tillbaka till akutmottagningen på grund av att stickningarna hade spridit sig upp till knänivå och det hade tillkommit stickningar också i fingrarna. Vid undersökning av neurologiskt status hade patienten stapplig gång, kunde stå på hälarna och tårna men tappade balansen vid tågång. Patellar- och akillesreflexer var svåra att utlösa, men biceps och brachioradialis var normala. Sensibiliteten i benen var något nedsatt, men sidlik. Övrigt neurologiskt status var normalt. Patienten skickades hem igen med rekommendation att söka vård vid försämring.
Han kom tillbaka till akutmottagningen samma dag på grund av att han inte kunde ta sig upp efter ett fall i hemmet. Han lades in på neurologavdelning med misstanke om Guillain–Barrés syndrom. Utredning med lumbalpunktion visade hög proteinnivå på 1 200 mg/l. Elektroneurografi visade sensomotorisk polyneuropati med demyeliniserande inslag i form av förlängning av distala latenser, avsaknad av F-svar (mått på nervens ledningsförmåga) och konduktionsblock, vilket stärkte misstanken om Guillain–Barrés syndrom. Intravenös immunglobulinbehandling administrerades i 5 dagar. Efter behandlingens avslut förbättrades motorisk funktion och patienten kunde stå på benen.
Några dagar senare flyttades han till rehabiliteringsavdelning för fortsatt träning, och i början av september skrevs han ut. Vid utskrivningen kunde patienten gå självständigt med stöd av 1–2 kryckor, alternativt rollator.
En vecka senare sökte patienten ånyo på grund av försämring av balans och styrka i övre och nedre extremiteter. Ny elektroneurografi visade uttalad generell sensomotorisk polyneuropati med tydlig progress. Patienten behandlades ånyo med intravenöst immunglobulin. Förbättringen varade bara i några dagar, därefter försämrades patienten åter. Han kunde inte stå på benen och hade svårt att förflytta sig från säng till rullstol. Försämringen fortskred och snart hade patienten svårt att röra på benen i sängen. Han hade nedsatt motorik och sensorik i sina händer, och man kunde se tydliga atrofier. Ny utredning påbörjades med kontroll av paramaligna antikroppar, DT torax och buk samt MR hjärna och helrygg. Samtliga undersökningar utföll normalt. Efter ytterligare en behandling med intravenöst immunglobulin och intravenöst kortison förbättrades patienten och han skrevs ut i början av december till korttidsboende med diagnosen CIDP. Vid utskrivningen klarade han självständiga förflyttningar till och från rullstol samt tränade gång med gåbord och uppresningar.
Förbättringen var dock snabbt övergående och en vecka senare var han så pass försämrad att han åter lades in. Behandlingen skärptes ytterligare, och förutom intravenöst immunglobulin och kortison erhöll han cyklofosfamid. Patienten svarade på denna behandling, och vid jultid kunde han klara förflyttningar själv med rullstol, och kraften i händerna förbättrades.
Trippelbehandlingen upprepades i början av januari, men redan i mitten av den månaden försämrades patientens status med tillkomst av sensoriska symtom i bålen utan centrala tecken i neurologiskt status. I slutet av januari utvecklade han en tetrapares, och han behövde hjälp med all aktivitet i dagliga livet (ADL). Den uttalade symtomprogressen föranledde multidisciplinär diskussion om att gå vidare mot autolog stamcellstransplantation, för vilken patienten accepterades.
Några dagar innan patienten skulle påbörja behandling med sikte mot autolog stamcellstransplantation drabbades han av heshet, andningssvårigheter, sväljningssvårigheter och paralys i alla extremiteter. Autolog stamcellstransplantation fick då blåsas av. Han flyttades till Iva för noninvasiv ventilation och 2 dygn senare till universitetssjukhus för plasmaferes. Elektroneurografi visade en tydlig progress med total avsaknad av registrerbara motoriska och sensoriska svar, och elektromyografi visade inga hållpunkter för generell sekundär axonal påverkan.
Efter 5 dagars plasmaferes och behandling med intravenöst immunglobulin och cyklofosfamid kunde man se en förbättring, som dock bara var tillfällig.
Patienten försämrades igen i sin andning och ådrog sig en lunginflammation, vilken ledde till respiratorvård. Han fick trakeostomi i mitten av februari och flyttades tillbaka till hemsjukhuset. Autonom dysfunktion med växlande blodtrycksfall och högt blodtryck krävde tät övervakning på Iva. Patienten erhöll fortsatt behandling med intravenöst kortison, och behandlingen intensifierades genom tillägg av rituximab. Lite senare drabbades han av en candidasepsis som behandlades med antimykotika med bra effekt. Under tiden utvecklade han en pancytopeni med LD-stegring på 150 mikrokat/l och ferritin på 33 000 mikrogram/l. PAD från kristabiopsi visade bild förenlig med hemafagocytos, vilken bedömdes orsakad av candidasepsis. Hemofagocytosen avtog på given behandling.
Patientens kliniska bild förbättrades drastiskt efter behandling med rituximab i kombination med intravenöst immunglobulin. Under april avvecklades sonden och trakeostomin. Han satt i rullstol och kunde äta själv. Under maj månad kunde han gå med gåbord och hade inget behov av hjälp vid ADL.
Behandlingsplanen är att han ska fortsätta med intravenöst immunglobulin och rituximab, och i nuläget planeras inte för autolog stamcellstransplantation.
Diskussion
Genomgång av bibliografi visar att 16 procent av CIDP-patienterna har akut eller subakut insjuknande som vid Guillain–Barrés syndrom. Dessa patienter kan ha bulbära symtom, autonom dysfunktion och andningssvikt. Denna typ av CIDP kallas akut insättande CIDP (A-CIDP). Elektrofysiologiska fynd vid Guillain–Barrés syndrom och A-CIDP kan vara lika vid insjuknandet. Kontinuerlig uppföljning kan hjälpa oss att skilja dem åt, vilket är viktigt med tanke på behandling och prognos (Tabell 1) [2].
Andningssvikt är sällsynt men kan också förekomma vid akut exacerbation hos patienter som har sin CIDP-diagnos sedan några månader eller år [3, 4]. Orsaken till andningssvikt vid Guillain–Barrés syndrom och CIDP är påverkan på andningsmuskulaturen. Svaghet i orofaryngeal muskulatur och pneumonier kan också påverka andningsfunktionen [5]. Demyelinisering i frenikusnerven och denervation i diafragma har beskrivits hos vissa CIDP-patienter som utvecklar andningssvikt [3-7]. Elektroneurografi kan vara av värde för bedömning av frenikusfunktionen. Elektromyografiundersökning samt ultraljud av diafragma kan påvisa paralys av diafragman [4, 5, 7, 8].
Hos vår patient fanns inga tecken till lunginflammation vid debut av andningssvikt. Progress av demyelinisering i extremiteter och svaghet i orofaryngeal muskulatur kunde vara orsaken till andningsinsufficiens. Man kunde inte utesluta påverkan av frenikusnerven och diafragma, då undersökning med elektroneurografi, elektromyografi och ultraljud inte genomfördes. Det har också beskrivits att påverkan av andningsmuskulaturen kan förekomma med eller utan uttalad tetrapares [2-6].
Det här fallet uppmärksammar oss på att patienter med CIDP som uppvisar tecken på ventilationspåverkan behöver tät övervakning med kontroll av andningsfrekvens, pulsoximetri och PEF (peak expiratory flow, topputandningsflöde), då ventilationsbehandling kan vara indicerad. Elektroneurografi av frenikusnerven och elektromyografi eller ultraljud av diafragma kan vara till hjälp för att identifiera riskpatienter i tid. Samtidigt behöver kardiella och pulmonella sjukdomar också uteslutas.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
FAKTA 1. Diagnostiska kriterier, Svenska neuromuskulära arbetsgruppen (SNEMA)
CIDP kliniska diagnoskriterier
Samtliga tre kriterier (A–C) bör uppfyllas:
A. Kronisk progressiv, stegvis eller intermittent proximal och distal muskelsvaghet och sensorisk påverkan i samtliga fyra extremiteter och eventuellt även kranialnerver.
B. Duration av den progressiva fasen >2 månader
C. Areflexi eller hyporeflexi i benen, samt eventuellt även i armarna.
CIDP elektrofysiologiska kriterier
Minst ett av följande (A–G):
A. Förlängning av distala motoriska latenser i två nerver
B. Reduktion av motorisk nervledningshastighet i två nerver
C. Förlängning av F-svarslatenser i två nerver
D. Avsaknad av F-svar i två nerver + en demyeliniserande parameter i >1 nerver
E. Partiell motorisk konduktionsblockad i två nerver eller i en nerv + minst en demyeliniserande parameter i >1 nerver.
F. Abnormal temporal dispersion i >1 nerver.
G. Distal CMAP-duration av minst 9 ms i en eller fler nerver + minst en annan demyeliniserande parameter i >1 nerver
CIDP stödjande kriterier
A. Stegrad cerebrospinal proteinnivå, samt antal celler <10/mm3.
B. MRI-undersökning som visar gadoliniumladdning och/eller hypertrofi av cauda equina-, lumbosakral- eller cervikala nervrötter, alternativt brakial- eller lumbosakralplexus.
C. Nervbiopsi med tydliga tecken på demyelinisering och/eller remyelinisering i >5 fibrer i elektronmikroskop, eller i >6 av 50 »teased fibres«.
D. Klinisk förbättring efter immunterapi.
Referenser
- Svenska neuromuskulära arbetsgruppen (SNEMA). CIDP konsensus SNEMA – diagnostik och behandling. http://www.snema.se/dokument_files/CIDP%20konsensus%20Diagnostik%20och%20behandling_2014.pdf
- Mansour M, Ouerdiene A, Bedoui I, et al. Acute-onset chronic inflammatory demyelinating polyneuropathy with cranial nerves and respiratory tract involvement: a case report. Clin Case Rep. 2020;8(11):2199-203.
- Henderson RD, Sandroni P, Wijdicks EFM. Chronic inflammatory demyelinating polyneuropathy and respiratory failure. J Neurol. 2005;252(10):1235-7.
- Zivkovic SA, Peltier AC, Iacob T, et al. Chronic inflammatory demyelinating polyneuropathy and ventilatory failure: report of seven new cases and review of the literature. Acta Neurol Scand. 2011;124(1):59-63.
- Kimber TE, Orrell RW, King RHM, et al. Pathological findings in a patient with ventilatory failure and chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Syst. 2003;8(1):13-6.
- Sanjeev JHA, Ansari MK, Sonkar KK, et al. Unusual features in chronic inflammatory demyelinating polyneuropathy: good outcome after prolonged ventilatory support. J Neurosci Rural Pract. 2011;2(2):171-3.
- Tataroğlu C, Özkul A, Şair A. Chronic inflammatory demyelinating polyneuropathy and respiratory failure due to phrenic nerve involvement. J Clin Neuromuscul Dis. 2010;12(1):42-6.
- Haji K, Butler E, Royse C. A case of chronic inflammatory demyelinating polyneuropathy with reversible alternating diaphragmatic paralysis: case study. Crit Ultrasound J. 2015;7(1):16.
- Internetmedicin.se; Vrethem M. Guillain–Barrés syndrom (GBS). 11 aug 2020. https://www.internetmedicin.se/behandlingsoversikter/neurologi/guillain-barres-syndrom-gbs/
- Press R, Askmark H, Andersen O. Inflammatoriska polyneuropatier kan behandlas framgångsrikt. Läkartidningen. 2012;109:950-4.
Summary
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired autoimmune inflammatory polyneuropathy characterized by a progressive or relapsing course. It has been reported that approximately 16% of CIDP patients may present with an acute clinical onset much like Guillain-Barré syndrome (GBS) but with a subsequent chronic progression. These patients are classified as acute-onset CIDP (A-CIDP). In CIDP, bulbar and ventilator involvement is uncommon.
We report a case of a 56-year-old man in previous good health who developed progressive paresthesia and weakness in the limbs within 2 weeks. The diagnosis of A-CIDP was made. The patient’s course fluctuated greatly over 5 months and he developed oropharyngeal weakness and ventilatory insufficiency that led to a tracheostomy. He slowly improved after IVIG treatment in combination with rituximab but required mechanical ventilatory support for 3 months. One month later he was able to perform all activities of daily living independently.


















