




VEXAS är ett autoinflammatoriskt syndrom som orsakas av somatiska mutationer i X-kromosomens UBA1-gen.
Tillståndet drabbar främst män och kännetecknas av vuxendebuterande och terapirefraktär inflammation med reumatologiska manifestationer och hematologiska rubbningar.
Gentest för UBA-1 ger diagnos.
Tillståndet kan svara bra på prednisolon i medelhöga doser, men ofta är det svårt att sänka dosen utan symtomrecidiv. Olika immundämpande läkemedel samt allogen stamcellstransplantation har prövats som behandling.
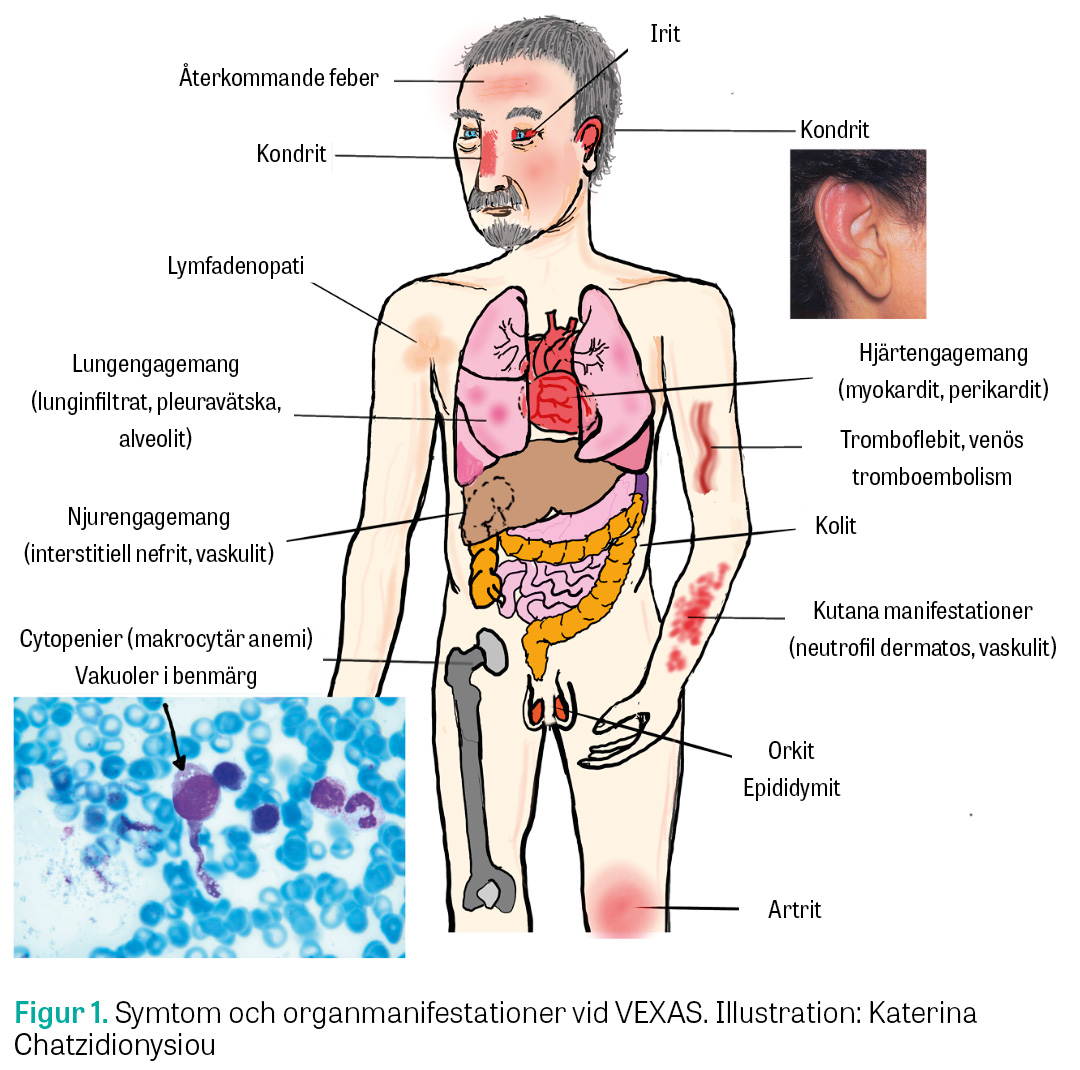
Ett autoinflammatoriskt syndrom kallat VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) beskrevs för första gången år 2020 och drabbar patienter med mutationer i X-kromosomens UBA1-gen [1]. Syndromet ger svårbehandlad inflammation och störd benmärgsfunktion. VEXAS drabbar främst äldre män och kan ge ett brett spektrum av symtom (Figur 1). Vi presenterar här de första fallbeskrivningarna av svenska patienter med VEXAS.
Fallbeskrivningar
Patient 1. En 72-årig man sökte vård på grund av B-symtom samt tromboflebit, men utredningen gav ingen klar diagnos. Följande år drabbades patienten av lungemboli och återkommande inflammatoriska symtom: dermatit, öronkondrit, irit, epididymit samt feber och lunginfiltrat. Antibiotika gav ingen symtomlindring, men prednisolon 20 mg/dag och azatioprin 100 mg/dag prövades, vilket initialt gav viss effekt. Patienten vårdades flera gånger med feber och andningssvikt och vid ett tillfälle krävdes intensivvård, då en RS-virusinfektion med bakteriell pålagring konstaterades. På grund av infektionsproblematik under kortisonbehandling och svårigheter att sänka dosen prövades en kortisonsparande behandling med metotrexat, anakinra och tocilizumab, dock utan tydlig effekt.
Patienten drabbades därefter av dyspné och feber under behandling med prednisolon, tocilizumab och metotrexat. Pulmonell Pneumocystisinfektion påvisades och en möjligen Listeriarelaterad exsudativ perikardit. Patienten behandlades med antibiotika, kolkicin och ökad dos prednisolon. Därefter beslutade man att avstå ytterligare immundämpande behandling på grund av tveksam effekt och infektionsproblematik. Vid sjukdomsskoven sågs kraftig inflammation och makrocytär anemi. Benmärgsprov visade granulocytär hyperplasi, hämmad erytropoes och lindriga dysplasier. Bilden bedömdes som reaktiv, och dysplasierna var otillräckliga för att diagnostisera myelodysplastiskt syndrom.
ANA, ANCA, anti-CCP samt genpanel för autoinflammatoriska tillstånd (33 gener) utföll negativa. Därefter utfördes tilläggsanalys av UBA1-genen som påvisade p.Met41Thr, en genetisk »hotspot«-mutation för VEXAS. I nuläget har patienten god effekt av prednisolon 15 mg/dag, men vid försök till dossänkning har han försämrats.
Patient 2. En 64-årig man inkom akut på grund av buksmärtor, trötthet och illamående. En portavenstrombos diagnostiserades och patienten behandlades med dalteparin. Blodstatus visade Hb 93 g/l, MCV 100 fL och TPK 105 × 109/l. Vidare påvisades en M-komponent, IgG-lambda 8 g/l. I benmärgsprov sågs en förhöjd cellhalt på 90 procent och signifikanta dysplasier förenliga med myelodysplastiskt syndrom. Dessutom sågs drygt 10 procent plasmaceller förenliga med multipelt myelom. Patientens karyotyp var normal och inga klonala mutationer kunde påvisas via genpanel.
Uppföljande benmärgsprov efter ett och ett halvt års understödjande behandling visade en morfologiskt oförändrad bild, men en ökning av p53-positiva celler, varför allogen stamcellstransplantation bedömdes indicerad. Azacitidin gavs för att reducera sjukdomsbördan inför transplantationen, dock med tveksam effekt. Transplantation med obesläktad donator genomfördes framgångsrikt och patienten är nu i remission, ett år efter transplantationen. I efterhand utfördes exomsekvensering i forskningssyfte, som påvisade en känd »hotspot«-mutation i UBA1, p.Met41Leu, och diagnosen VEXAS ställdes.
Patofysiologi: genetiska förändringar
VEXAS orsakas av förvärvade (somatiska) varianter i UBA1-genen som är belägen på X-kromosomen (Xp11.3) [1]. Män har enbart en kopia av UBA1-genen, vilket förklarar varför främst män drabbas, även om ett fåtal kvinnor med VEXAS har beskrivits [2]. UBA1-genen kodar enzymet E1, som aktiverar det första steget i ubikvitinering av proteiner, en regleringsprocess för nedbrytning av dessa. Medfödda varianter i UBA1-genen har beskrivits hos pojkar med en sällsynt X-bunden form av spinal muskelatrofi, med hypotoni och medfödda ledfelställningar [3]. De varianter som påvisats vid VEXAS är i stället förvärvade och har detekterats i blodceller, främst i hematopoetiska stamceller och utmognande myeloiska celler [1]. VEXAS är således inte ett ärftligt tillstånd.
Klinisk bild
Flera gemensamma symtom beskrevs i den första VEXAS-kohorten, bland annat polykondrit, feber och lunginfiltrat [1]. Därefter har fler symtom kompletterat den kliniska bilden (Figur 1) [4-6]. De vanligaste symtomen i en stor fransk VEXAS-kohort (116 patienter) var dermatos (83 procent), feber (65 procent) och viktnedgång (54 procent). 35 procent drabbades av oprovocerad trombos. Hög mortalitet observerades: efter 3 års medianuppföljning hade 15 procent av patienterna avlidit. Gastrointestinalt engagemang, lungengagemang och förstorade mediastinala lymfkörtlar beskrevs som faktorer associerade med död [6]. Ett fåtal fall av kvinnliga VEXAS-patienter finns beskrivna [2], och i den franska kohorten var 4 procent kvinnor [6]. Medianåldern för insjuknande har estimerats till 64–74 år [1, 2, 4, 6, 7], men enstaka fall har rapporterats hos patienter i 40-årsåldern [8]. Många patienter med VEXAS uppfyller även kriterierna för något av de beskrivna associerade syndromen (Fakta 1) [1].
Diagnostik
Typiska symtom hos män över 40 år bör inge misstanke om VEXAS (Fakta 2). Tidig diagnostik minskar risken för komplikationer orsakade av onödiga invasiva diagnostiska procedurer eller ineffektiva tungt immunsuppressiva läkemedel. Inga sjukdomsspecifika antikroppar har identifierats, men RF, lupusantikoagulans eller ANA kan ibland påvisas. CRP och SR är i regel förhöjda under sjukdomsskov [1].
Hematologiska rubbningar vid VEXAS utgörs av vakuoler i erytropoetiska och myelopoetiska celler i benmärgen, tromboembolism samt myelodysplastiskt syndrom och multipelt myelom/monoklonal gammopati av oklar signifikans. Myelodysplastiskt syndrom har rapporterats hos 25–55 procent av patienterna med VEXAS [1, 7, 9] och multipelt myelom/monoklonal gammopati av oklar signifikans hos cirka 20 procent [1]. VEXAS kan dock vara associerat med cytopenier och makrocytos utan att uppfylla patomorfologiska kriterier för myelodysplastiskt syndrom. Vakuoler i erytropoetiska/myelopoetiska celler förekommer även vid myelodysplastiskt syndrom utan VEXAS, vilket försvårar diagnostiken [10]. Det är lämpligt att dessa patienter utreds via ett multidisciplinärt team, som åtminstone består av reumatolog, hematolog och klinisk genetiker.
Vid misstanke om VEXAS utreds patienten med benmärgsprov och genanalys. Som genetisk metod bör någon form av DNA-sekvensering av UBA1-genen användas. De flesta patienter med VEXAS har så kallade missense-varianter i UBA1 som påverkar aminosyran metionin i position 41 av proteinet, men även andra somatiska varianter har rapporterats och därför rekommenderas en fullständig sekvensering av genens kodande delar [11]. Troligen kommer UBA1 inom ett år att ingå i genpaneler för utredning av myeloiska maligniteter och autoinflammatoriska tillstånd. Fram till dess bör diskussion kring metodval ske med närmaste enhet för klinisk genetik. Påvisandet av en förvärvad, och tidigare beskriven, sekvensvariant för VEXAS räcker för att ställa diagnos.
Behandling
Ingen riktigt effektiv behandling mot VEXAS har hittills identifierats. Kortison fungerar ofta bra, men relativt höga doser prednisolon (15–20 mg/dag) kan behövas under lång tid [4, 6] vilket ger hög biverkningsrisk. I en retrospektiv studie [7] utvärderades olika kortisonsparande läkemedel. Den tydligaste antiinflammatoriska effekten sågs med tocilizumab, ciklosporin och januskinashämmare. Man bör dock tolka resultaten med försiktighet med tanke på den retrospektiva designen och korta uppföljningstiden. Med dagens kunskapsläge är allogen stamcellstransplantation den enda potentiellt botande behandlingen [4, 8]. På grund av behandlingsrelaterade risker vid stamcellstransplantation behövs en noggrann selektion av lämpliga patienter.
Konklusion
VEXAS är ett nyupptäckt genetiskt syndrom med överlappande reumatologiska manifestationer och hematologiska rubbningar. På grund av den varierande kliniska bilden är det viktigt att läkare i alla delar av vården känner till syndromet. Då mortaliteten är hög behövs randomiserade studier för att utvärdera olika behandlingsstrategier.
Läs författarintervju:
4 frågor: Ett syndrom som expanderar vår kunskap om genetik
Potentiella bindningar eller jävsförhållanden: Karin Gunnarsson har erhållit arvode från Boehringer Ingelheim för en föreläsning år 2020.
Monika Klimkowska, klinisk patologi, har bidragit med granskning av benmärgsprov och bild på vakuoler.
FAKTA 1. Associerade sjukdomar och syndrom [1]
- Recidiverande polykondrit
- Jättecellsarterit
- Polyarteritis nodosa
- Akut febril neutrofil dermatos (Sweets syndrom)
- Myelodysplastiskt syndrom
- Multipelt myelom
FAKTA 2. VEXAS bör misstänkas vid en kombination av följande symtom
- Oklar och behandlingsresistent inflammation och feber hos män över 40 års ålder
- Inflammatoriska symtom, till exempel polykondrit, vaskulit, hudutslag, lunginfiltrat och artriter
- Hematologiska manifestationer, till exempel tromboembolism, makrocytär anemi och persisterande cytopenier
Referenser
- Beck DB, Ferrada MA, Sikora KA, et al. Somat-ic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383(27):2628-38.
- Barba T, Jamilloux Y, Durel CA, et al. VEXAS syndrome in a woman. Rheumatology (Oxford). 2021;60(11):e402-3.
- Ramser J, Ahearn ME, Lenski C, et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atro-phy. Am J Hum Genet. 2008;82(1):188-93.
- Koster MJ, Kourelis T, Reichard KK, et al. Clin-ical heterogeneity of the VEXAS syndrome: a case series. Mayo Clin Proc. 2021;96(10):2653-9.
- van der Made CI, Potjewijd J, Hoogstins A, et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: a Dutch case series of patients with VEXAS. J Allergy Clin Immunol. 2022;149(1):432-9.e4.
- Georgin-Lavialle S, Terrier B, Guedon AF, et al; GFEV, GFM, CEREMAIA, MINHEMON. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. 2022;186(3):564-74.
- Bourbon E, Heiblig M, Gerfaud Valentin M, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. 2021;137(26):3682-4.
- Diarra A, Duployez N, Fournier E, et al. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a two center experience. Blood Adv. 2022;6(3):998-1003.
- Poulter JA, Collins JC, Cargo C, et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood. 2021;137(26):3676-81.
- Gurnari C, Pagliuca S, Durkin L, et al. Vacuolization of hematopoietic precursors: an enigma with multiple etiologies. Blood. 2021;137(26):3685-9.
- Comont T, Heiblig M, Rivière E, et al; French VEXAS study group; Groupe Francophone des Myélodysplasies (GFM); Medecine interne, hémato et onco (MINHEMON) group. Azacitidine for patients with vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. 2022;196(4):969-74.
Summary
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) is a newly discovered syndrome caused by a somatic mutation in the UBA1 gene, located in the X chromosome. The syndrome mainly affects older men, and presents with persistent inflammation and rheumatological symptoms like polychondritis, lung infiltrates and dermatitis. Related hematological disturbances are thromboembolic events, macrocytic anemia, myelodysplastic syndrome, and vacuoles found in bone marrow hematopoietic cells. A genetic test of the UBA1 gene confirms the diagnosis when a clinical suspicion of VEXAS is raised. Patients usually respond to prednisolone at a dose of 15-20 mg/day but an effective and well tolerated long-term treatment strategy is still to be defined. The only potentially curative treatment is allogeneic stem cell transplantation. In this case report we present two cases of VEXAS, one of which has undergone an allogeneic stem cell transplantation.


















