Pulmonell hypertension är en vanlig komplikation till vänstersidig hjärtsjukdom och medför såväl försämrad livskvalitet som försämrad prognos.
Den pulmonella hypertensionen beror initialt på passiv stockning av blod i lungkretsloppet men kan kompliceras av såväl pulmonell vasokonstriktion som vaskulär remodulering.
2015 års europeiska riktlinjer innefattar en ny definition av pulmonell hypertension sekundär till vänstersidig hjärtsjukdom, vilken tagits fram för att möjliggöra adekvat subgruppering av tillståndet.
Specifik medicinsk behandling för de prekapillära förändringarna i lungcirkulationen saknas. Terapin syftar i stället till att optimera bakomliggande hjärtsjukdom.
Vänstersidig hjärtsjukdom innefattar en rad olika tillstånd, av vilka vänstersidig hjärtsvikt utgör den absolut största patientgruppen. Prevalensen för hjärtsvikt har uppskattas till cirka 2 procent med en tydlig ökning i högre åldrar [1]. När hjärtat sviktar och ej förmår upprätthålla adekvat hjärtminutvolym sker en aktivering av neurohormonella system, inklusive renin–angiotensin–aldosteronsystemet och det sympatiska nervsystemet. Långvarig neurohormonell aktivering resulterar i salt- och vätskeretention samt ökad systemvaskulär resistans med förhöjda vänstersidiga fyllnadstryck och progredierande vänsterkammardysfunktion som följd [2]. Vid vänstersidig hjärtsjukdom är pulmonell hypertension (PH) en komplicerande faktor vilken drabbar en majoritet av patienterna någon gång under sjukdomsförloppet och medför såväl försämrad livskvalitet som försämrad prognos [3-5]. Symtomen vid pulmonell hypertension på basen av vänstersidig hjärtsjukdom, ofta betecknad med sin engelska förkortning PH-LHD, är icke-specifika men innefattar, liksom okomplicerad vänstersidig hjärtsjukdom, dyspné. Detta medför svårigheter att kliniskt särskilja patienter med PH-LHD från dem utan pulmonell hypertension.
Pulmonell hypertension delas in i fem subgrupper baserat på underliggande sjukdom (Fakta 1) [3]. Grupp I (pulmonell arteriell hypertension), grupp III (pulmonell hypertension på basen av lungsjukdom), grupp IV (kronisk tromboembolisk pulmonell hypertension) och grupp V (pulmonell hypertension på basen av okända eller multifaktoriella mekanismer) definieras samtliga som pre-kapillära, medan grupp II, det vill säga PH-LHD, definieras som postkapillär. Under de senaste decennierna har ett växande vetenskapligt intresse för pulmonell arteriell hypertension och kronisk tromboembolisk pulmonell hypertension resulterat i ökad förståelse och etablering av nya behandlingsstrategier för dessa tillstånd. I jämförelse är den patofysiologiska kunskapen kring PH-LHD bristande, och specifika behandlingar för detta tillstånd saknas. Detta trots att PH-LHD är den helt dominerande gruppen och att pulmonell hypertension är en negativ prognostisk faktor vid vänstersidig hjärtsjukdom [3-5].
I början av 2016 publicerade de europeiska kardiolog- och lungmedicinska förbunden nya riktlinjer för pulmonell hypertension [3]. I dessa presenterades såväl nya utredningsmodeller som en ny klassifikation av PH-LHD. Då en god förståelse för innebörden av PH-LHD är av stor vikt för alla specialister som behandlar patienter med vänstersidig hjärtsjukdom presenterar vi här, utifrån nya riktlinjer, aktuella kunskaper om patofysiologiska och patobiologiska mekanismer, epidemiologi samt utredning och behandling av detta tillstånd.
Definition
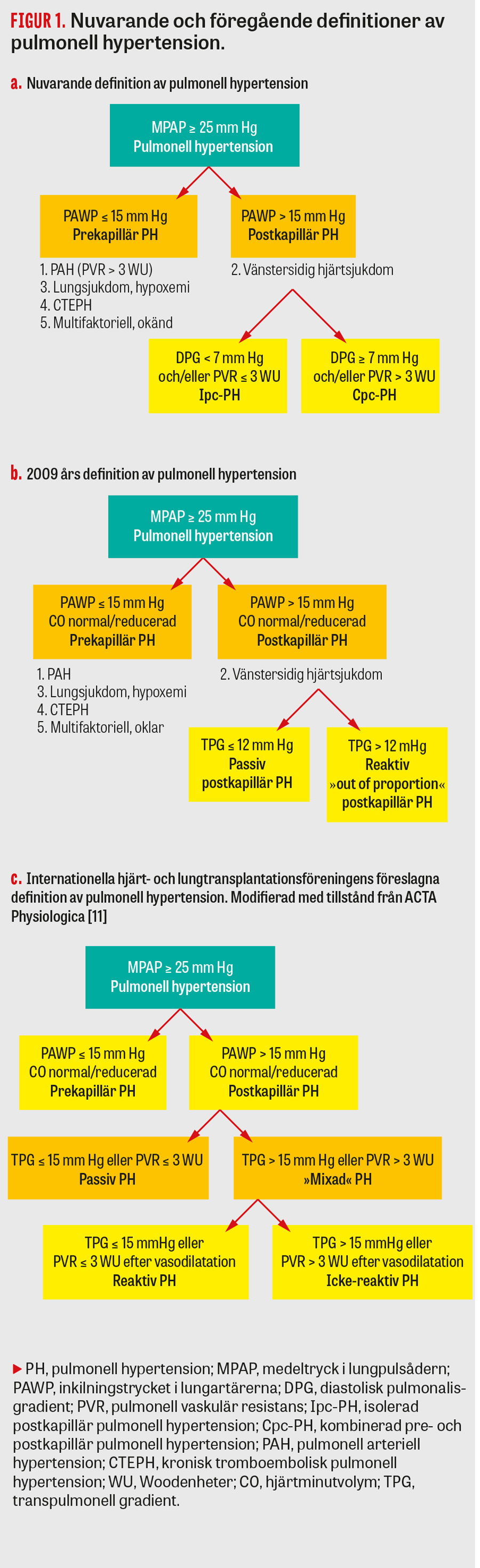
I den kliniska vardagen upptäcks vanligen pulmonell hypertension med ekokardiografi och diagnostiseras med högersidig hjärtkateterisering. Ekokardiografiskt anses en trikuspidalis insufficiens med maximal hastighet > 2,8 m/s vara det mått som starkast inger misstanke om pulmonell hypertension [3]. Via omräkning med hjälp av Bernoullis ekvation och tillägg av trycket i vena cava inferior motsvarar detta ett systoliskt tryck i arteria pulmonalis > 35 mm Hg. Det finns dock ett flertal felkällor vid ekokardiografi, och vid misstanke om annan orsak till pulmonell hypertension än vänstersidig hjärtsjukdom, såsom pulmonell arteriell hypertension, kronisk tromboembolisk pulmonell hypertension och shunt samt vid utredning för hjärttransplantation, ska pulmonell hypertension bedömas invasivt med högersidig hjärtkateterisering. Denna utförs vanligen genom introduktion av en tryckmätningskateter via vena jugularis interna alternativt vena femoralis till höger hjärthalva. Vid undersökningen mäts trycken i höger förmak och höger kammare samt i arteria pulmonalis. Man mäter även inkilningstrycket i lungartärerna (PAWP, pulmonary artery wedge pressure), som utgör ett indirekt mått på trycket i vänster förmak, och hjärtminutvolymen beräknas genom så kallad termodilution eller Fick-mätningar. Vid högersidig hjärtkateterisering definieras pulmonell hypertension som ett medeltryck i arteria pulmonalis ≥ 25 mm Hg och subgrupperas sedan i en prekapillär och en postkapillär grupp beroende på nivån av PAWP [3]. Vid postkapillär pulmonell hypertension, det vill säga PH-LHD (grupp II) är PAWP > 15 mm Hg, medan PAWP är ≤ 15 mm Hg i grupp I, III, IV och V, vilka således definieras som prekapillära [3].
Enligt nya riktlinjer subgrupperas PH-LHD i en isolerad postkapillär grupp (Ipc-PH), med låg pulmonell vaskulär resistans, PVR ([medeltryck i arteria pulmonalis – PAWP]/hjärtminutvolym) och låg diastolisk pulmonalisgradient, DPG (diastoliskt tryck i arteria pulmonalis – PAWP), samt i en kombinerad pre- och postkapillär grupp (Cpc-PH) med höga PVR- och DPG-nivåer (Figur 1a) [3]. Denna definition är justerad jämfört med tidigare versioner av riktlinjer (Figur 1b) i ett försök att adekvat särskilja patienter med prekapillära funktionella och/eller strukturella förändringar från dem med enbart passivt förhöjda tryck, sekundära till den vänstersidiga hjärtsjukdomen. Anledningen till att man valt att basera subgrupperingen på kombinationen av PVR och DPG är att DPG visat sig vara mindre beroende av flöde och av PAWP än den tidigare använda transpulmonella gradienten [6]. Normalvärde för medeltrycket i arteria pulmonalis anses vara 14 mm Hg och väldigt få friska individer bedöms ha ett medeltryck över 20 mm Hg, medan betydelsen av ett medeltryck i arteria pulmonalis 21–24 mm Hg är oklar [2]. För PVR och DPG uppskattas normalvärden till 1 WU (Woodenheter) [2] respektive 1–3 mm Hg [4]. Nivåerna varierar dock i litteraturen, och kliniskt anses vanligen PVR ≤ 1,7 WU normalt. Faktorer som övervätskning och hypoxi kan dessutom orsaka temporära förändringar av fyllnadstrycken och därmed hemodynamiken och därigenom komplicera subklassificering av den enskilda individen. Vidare har de olika parametrarna specifika brister som ytterligare försvårar bedömningen. Bland dessa kan nämnas att PVR påverkas av slagvolym, flöden och tryck i vänster förmak [6] medan DPG påverkas av hjärtfrekvensen [7]. Med anledning av dessa begränsningar är den nya subklassificeringen av PH-LHD fortsatt omdebatterad [8-10].
Patobiologi och patofysiologi
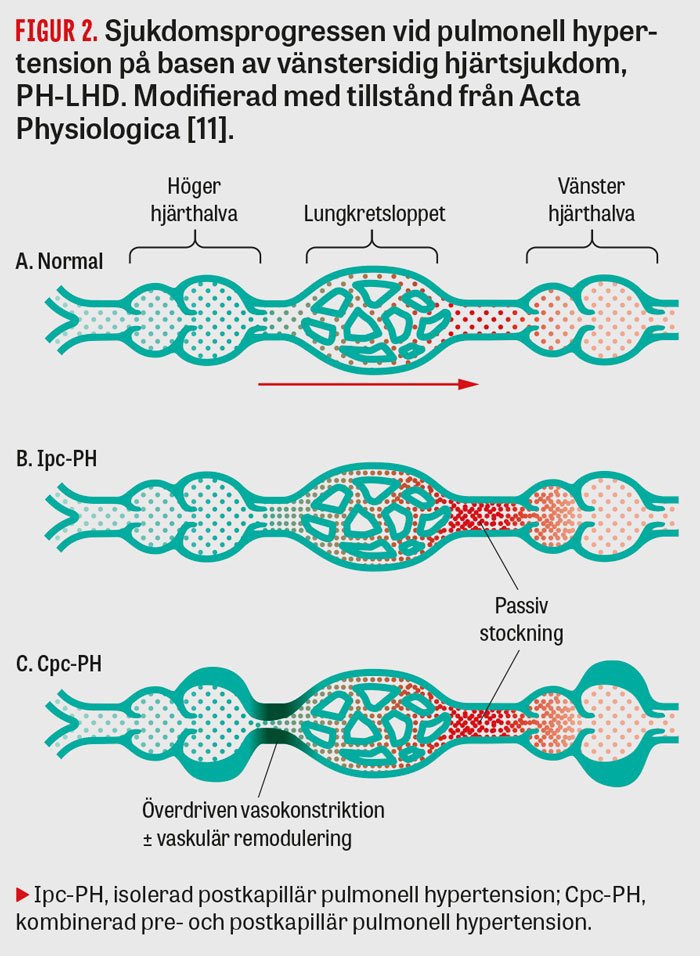
Den definitiva patofysiologin vid PH-LHD är okänd och sannolikt multifaktoriell. Det är dock välkänt att nedsatt vänsterkammarfunktion, med normal eller reducerad ejektionsfraktion, klaffvitier eller andra mindre vanliga orsaker till PH-LHD, exempelvis kongenitala hjärtfel, kan resultera i förhöjda tryck i vänster förmak som i sin tur fortplantas bakåt i lungkretsloppet. Denna passiva stockning av blod bakåt i lungkretsloppet, med förhöjt tryck i arteria pulmonalis som följd, benämns i nuvarande riktlinjer som Ipc-PH och utgör den första fasen av PH-LHD. I detta stadium förblir PVR och DPG, med reservation för de potentiella felkällor som beskrivs i tidigare stycken, låga (Figur 2) [11].
I akutskedet kan det förhöjda trycket ge upphov till kapillärskada och efterföljande ödem, vilka är reversibla [12, 13]. Om tryckstegringen kvarstår under en längre tid kan den däremot resultera i kvarstående endotelskada och endoteldysfunktion [14,15]. Endotelskadan leder i sin tur till en obalans mellan vasoaktiva substanser, inklusive nedsatt produktion av vasodilaterande ämnen som kvävemonoxid och ökad mängd endotelin, en potent vasokonstriktor [14-17]. Obalansen resulterar i en inadekvat relaxation av glatta muskelceller med överdriven vasokonstriktion som följd, vilket hemodynamiskt speglas av ökade PVR- och DPG-nivåer, så kallad Cpc-PH (Figur 2) [3].
Initialt är även denna vasokonstriktion i de flesta fall reversibel med vasodilaterande läkemedel, vilket vid hjärtkateterisering belyses med exempelvis nitroprussidinfusion. För att säkerställa att lungkärlsresistansen ej är fixerat förhöjd genomförs sådan infusion bland annat som provokationstest i samband med hjärttransplantationsutredningar [18]. I vissa fall kan dock en långvarig konstriktion ytterligare kompliceras av kroniska förändringar, inklusive remodulering, av lungkärlen [19]. Dessa förändringar liknar delvis dem man ser vid pulmonell arteriell hypertension och innefattar såväl intima fibros som media hypertrofi (Figur 2). De för pulmonell arteriell hypertension typiska plexiforma lesionerna tycks dock saknas vid PH-LHD.
Den nuvarande hemodynamiska definitionen av PH-LHD saknar kriterier för när vasodilaterande provokationstest ska genomföras, och det är därför svårt att adekvat differentiera mellan den reversibla vasokonstriktionen och den fixerade vaskulära remoduleringen. Ur detta perspektiv har Internationella hjärt- och lungtransplantationsföreningen (ISHLT, International Society for Heart and Lung Transplantation) föreslagit egna rekommendationer [18] för när dessa vasodilatationstest ska genomföras (Figur 1c). På grund av bristande evidens och avsaknad av normalvärden har dessa dock ej implementerats i de aktuella europeiska riktlinjerna [3]. Utöver avsaknaden av en hemodynamisk definition för att tydligt särskilja mellan de prekapillära förändringarna saknas även icke-invasiva metoder för att subgruppera PH-LHD. Detta är ett problem, då Cpc-PH med vaskulär remodulering är ett särskilt allvarligt tillstånd som kan orsaka akut högersidig hjärtsvikt efter hjärttransplantation, då det donerade hjärtats högerkammare ej förmår pumpa mot den kvarstående förhöjda resistansen i lungkretsloppet [18]. Om remodulering av lungkärlen är fixerad och ej påverkas av vasodilaterande farmaka kan detta alltså utgöra en direkt kontraindikation för hjärttransplantation [18]. Framtida arbeten krävs därför för att ta fram tydligare invasiva, såväl som icke-invasiva, metoder för diagnos och subklassificering av PH-LHD, för att på så vis öka förståelsen för detta tillstånd, optimera överlevnaden efter hjärttransplantation och möjligen identifiera nya behandlingar.
Epidemiologi
Liksom vid vänstersidig hjärtsjukdom är de vanligaste orsakerna till PH-LHD systolisk (HFrEF, hjärtsvikt med nedsatt ejektionsfraktion) och diastolisk (HFpEF, hjärtsvikt med bevarad ejektionsfraktion) hjärtsvikt. Vidare utgörs en mindre del av PH-LHD av klaffvitier och kongenital hjärtsjukdom [3]. Det tål att poängteras att prevalensen av PH-LHD skiljer sig åt beroende på bakomliggande hjärtsjukdom och dess svårighetsgrad. Det är också svårt att presentera exakta siffror avseende prevalensen då de epidemiologiska studier som genomförts använt såväl olika mätmetoder (vanligen hjärtkateterisering eller ekokardiografi) som olika kriterier för att definiera pulmonell hypertension. Med anledning av ovan nämnda faktorer har prevalensen i olika studier varierat mellan 25 och drygt 80 procent. PH-LHD är således mycket vanligt vid vänstersidig hjärtsjukdom, och en sammantagen bedömning är att prevalensen av pulmonell hypertension vid vänstersidig hjärtsjukdom är över 50 procent [20]. En majoritet har Ipc-PH [21], men andelen med Cpc-PH ökar med sjukdomens svårighetsgrad [21-23]. Även prevalensen av Ipc-PH respektive Cpc-PH är osäker och varierar mellan 12 och 38 procent beroende på om grupperingen baseras på DGP eller PVR, med betydligt högre prevalens när PVR används [5, 24]. Hur stor del av patienterna med Cpc-PH som har en fixerad pulmonell hypertension med uttalad vaskulär remodulering är heller ej känt.
En adekvat prevalensbedömning av subgrupperna försvåras också av att de flesta studier baserats på ekokardiografiska data med surrogatmått och begränsade möjligheter för exakta mätningar av flöden och tryck [3, 20]. Jämfört med patienter med pulmonell arteriell hypertension är de med PH-LHD äldre och har större andel kardiovaskulära komorbiditeter. De lider även i större utsträckning av metabola syndromet. Angående riskfaktorer för Cpc-PH har ett ekokardiografiskt mått, nämligen AV-planets rörelse mätt över höger kammares fria vägg (TAPSE) dividerat med systoliskt tryck i arteria pulmonalis, visats vara en oberoende prediktor vid pulmonell hypertension på basen av systolisk hjärtsvikt [21]. Vidare har ålder, klaffsjukdom och TAPSE/systoliskt tryck i arteria pulmonalis samtliga visat sig vara oberoende prediktorer för pulmonell hypertension orsakad av diastolisk hjärtsvikt, snarare än pulmonell arteriell hypertension [21].
Utredning
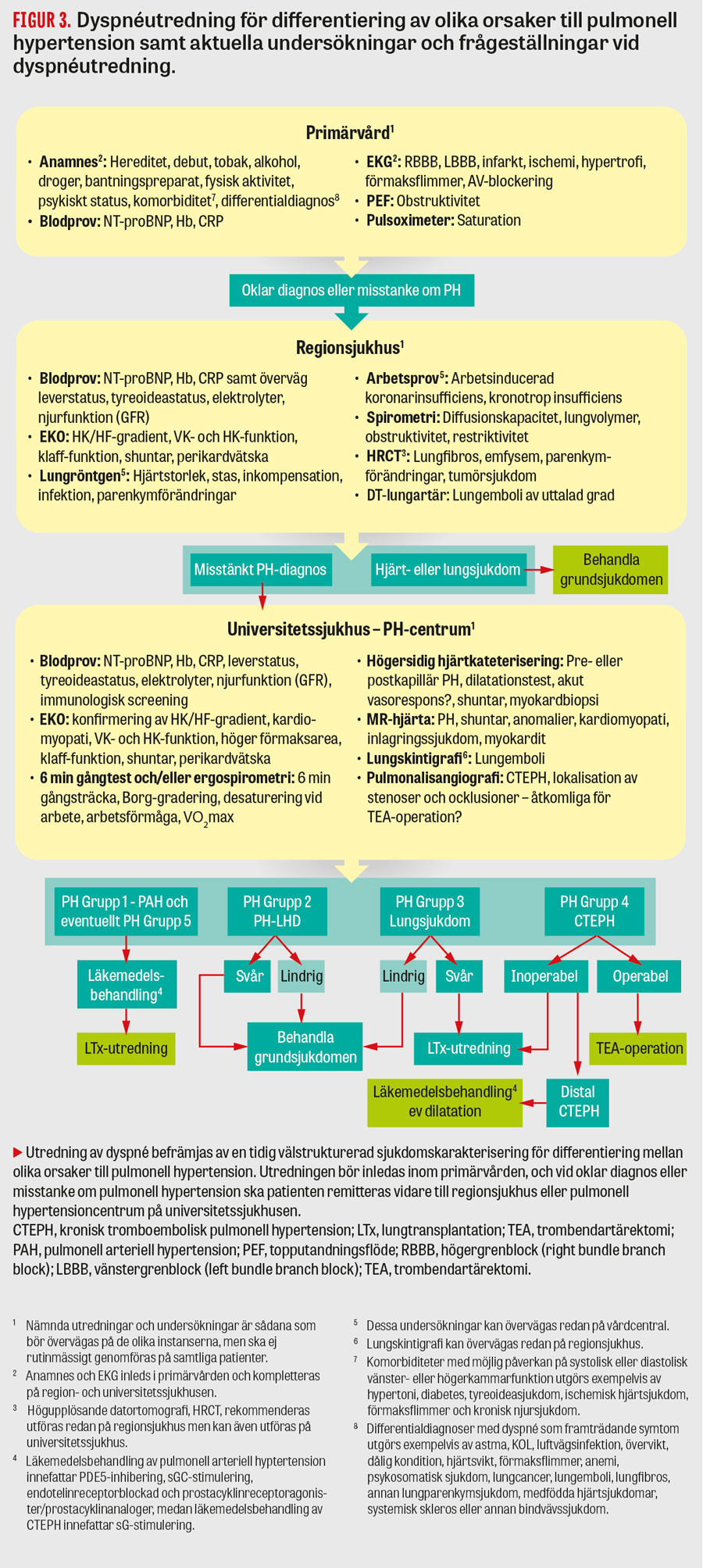
Förutom att identifiera en vänstersidig hjärtsjukdom, inklusive hjärtsvikt, är det primära syftet med utredning av patienter med uttalad dyspné och misstänkt pulmonell hypertension att utesluta andra, behandlingsbara orsaker till patientens tillstånd. Sådana orsaker inkluderar bland annat pulmonell arteriell hypertension och såväl akut som kronisk lungembolisering. Tydliga riktlinjer för hur svår kronisk dyspné och misstänkt PH-LHD ska utredas saknas, men det råder konsensus kring att sådan utredning, förutom klinisk bedömning, ska innefatta ekokardiografisk undersökning och EKG samt eventuellt ytterligare bilddiagnostiska undersökningar såsom MR, lungskintigrafi och högupplösande datortomografi samt spirometri [3]. Ålder > 65 år, tecken på vänstersidig hjärtsjukdom, metabola syndromet, förmaksflimmer och ischemisk hjärtsjukdom samt avsaknad av högerkammardysfunktion och perikardvätska är några tecken som talar för PH-LHD snarare än prekapillär pulmonell hypertension. Utifrån nuvarande riktlinjer för pulmonell hypertension och översiktsartiklar inom området [3, 4, 25] presenterar vi här ett förslag för en algoritm för utredning av kronisk dyspné och misstänkt pulmonell hypertension (Figur 3). En sådan utredning ska dock anpassas efter varje individs sjukdomsbild. Samtliga patienter behöver således ej bli föremål för alla föreslagna undersökningar.
Invasiv hemodynamisk undersökning med högersidig hjärtkateterisering rekommenderas i de fall då det råder klar misstanke om pulmonell arteriell hypertension eller kronisk tromboembolisk pulmonell hypertension, samt då orsaken till pulmonell hypertension är oklar eller om patienten är refraktär för behandling riktad mot grundsjukdomen, samt för de patienter med PH-LHD som är under utredning för hjärttransplantation. I det sistnämnda fallet är högersidig hjärtkateterisering särskilt viktig för att identifiera patienter med hög risk för akut högersidig hjärtsvikt tidigt efter transplantation [3, 18]. För att minska risken för svårtolkade och felaktiga resultat vid hjärtkateterisering ska patienters vätskestatus, i de fall det är möjligt, optimeras före undersökning.
Prognos
Pulmonell hypertension är en negativ prognostisk markör vid vänstersidig hjärtsjukdom [4]. Flera studier visar att den ökade dödligheten gäller vid hjärtsvikt med såväl bevarad [26] som nedsatt ejektionsfraktion [27] och att överlevnaden i dessa populationer är sämre ju högre medeltrycket i arteria pulmonalis är. [22]. Vidare visade en studie nyligen att överlevnaden för patienter med pulmonell arteriell hypertension och PH-LHD sekundär till diastolisk dysfunktion inte skiljer sig åt mellan de båda grupperna [28]. Slutligen är mortaliteten högre vid Cpc-PH än Ipc-PH [5,24], men det debatteras ännu vilken hemodynamisk markör som har starkast prognostiskt värde och det har publicerats motstridiga resultat kring hur såväl DPG som PVR är relaterat till överlevnaden vid PH-LHD [5, 8-10, 24].
Behandling
Specifik behandling för den pulmonella komponenten vid PH-LHD saknas. Vasodilaterande läkemedel med indikationen pulmonell arteriell hypertension och kronisk tromboembolisk pulmonell hypertension, innefattande fosfodiesteras typ 5-hämmare, guanylatcyklasstimulerare, endotelinreceptorblockerare och prostacyklinanaloger har med delvis positiva resultat undersökts i ett flertal mindre studier av vänstersidig hjärtsjukdom [11]. Dessa resultat har dock inte kunnat replikeras i större, kontrollerade och randomiserade studier, och behandling med prostacyklinanaloger har till och med fått avbrytas i förtid på grund av en trend mot ökad mortalitet hos hjärtsviktspatienter som givits aktivt läkemedel [29]. I detta sammanhang bör det poängteras att många av de tidigare studierna ej tydligt selekterat patienter med Cpc-PH utan även inkluderat patienter med Ipc-PH och i många fall även vänstersidig hjärtsjukdom utan pulmonell hypertension. Det är möjligt att utfallet varit annorlunda om endast patienter med ökad pulmonell vasokonstriktion, med eller utan vaskulär remodulering, inkluderats.
För närvarande pågår studier med fosfodiesteras typ 5-hämmare, endotelinreceptorblockerare och guanylatcyklasstimulerare vid PH-LHD, där den senare visat sig ha gynnsamma effekter på NT-proBNP vid vänstersidig hjärtsvikt [30]. Sammantaget saknas dock evidens för behandling med dessa så kallade pulmonell arteriell hypertension-specifika läkemedel vid PH-LHD, och i väntan på resultat från pågående kliniska läkemedelsstudier rekommenderas därför inte sådan behandling.
I stället fokuserar behandlingen i dag på att optimera underliggande sjukdom samt vätskestatus och innefattar utöver medikamentell terapi även kirurgisk åtgärd av exempelvis klaffsjukdom och medfödda hjärtvitier. Skulle denna behandling ej visa sig tillräcklig kan mekanisk vänsterkammarassist övervägas i noggrant utvalda fall av PH-LHD med välfungerande högerkammare, antingen för livslång behandling, så kallad »destinationsterapi«, vilket i Sverige endast genomförs inom ramen för en kontrollerad randomiserad studie (SweVAD), eller i väntan på hjärttransplantation, så kallad »brygga till transplantation«. Långtidsavlastning av vänster kammare med mekanisk hjärtpump har visats kunna reversera Cpc-PH och därigenom göra hjärttransplantation möjlig utan ökad risk för postoperativ högerkammarsvikt [31, 32]. Patienter med Ipc-PH har också kunnat genomgå hjärttransplantation med lika gott postoperativt resultat som patienter utan preoperativ PH-LHD, och ur perspektivet pulmonell hypertension är dessa således ej i behov av mekanisk vänsterkammarassist [31].
Sammanfattning
Pulmonell hypertension är ett vanligt, allvarligt och komplicerande tillstånd vid vänstersidig hjärtsjukdom. Trots detta är kunskapsläget dåligt, och med nuvarande metoder är det svårt att adekvat subgruppera PH-LHD. Detta är av betydelse, då Cpc-PH har visat sig medföra sämre prognos än Ipc-PH samt eftersom Cpc-PH kan försämra överlevnaden i samband med den ultimata hjärtsviktsbehandlingen, hjärttransplantation. Vidare saknas riktad behandling mot den prekapillära komponenten av PH-LHD. Läkemedel specifika för pulmonell arteriell hypertension och kronisk tromboembolisk pulmonell hypertension rekommenderas därför ej vid PH-LHD. I stället fokuserar behandlingen på att optimera bakomliggande sjukdom, och i enstaka fall, när detta inte är tillräckligt, kan mekanisk hjärtpump och/eller hjärttransplantation övervägas.
Sammantaget är framtida kliniska studier med noggrann patientselektion nödvändiga för att öka vår förståelse för PH-LHD och på så vis identifiera bättre metoder för diagnostik och behandling av detta vanliga och allvarliga tillstånd.
Potentiella bindningar eller jävsförhållanden: Jakob Lundgren har mottagit föreläsningsarvode från Actelion Pharmaceuticals Sverige AB och GlaxoSmithKline utanför det aktuella arbetet samt erhållit forskningsstipendium från Svensk förening för pulmonell hypertension och Actelion Pharmaceuticals Sverige AB.
Göran Rådegran har mottagit föreläsningsarvode från Actelion Pharmaceuticals Sverige AB, Bayer Healthcare, GlaxoSmithKline, NordicInfu Care och Sandoz/Novartis utanför det aktuella arbetet samt erhållit forskningsstipendium från Svensk förening för pulmonell hypertension, Actelion Pharmaceuticals Sverige AB samt GlaxoSmithKline. Göran Rådegran är, och har varit, huvud- eller biprövare i kliniska studier av pulmonell arteriell hypertension för GlaxoSmithKline, Actelion Pharmaceuticals Sverige AB, Pfizer, Bayer Healthcare och United Therapeutics samt i kliniska hjärttransplantationsstudier för Novartis.
Fakta 1. Övergripande klassifikation av pulmonell hypertension
I. Pulmonell arteriell hypertension
I’. Pulmonell venocklusiv sjukdom och/eller pulmonell kapillär hemangiomatos
I’’. Persisterande pulmonell hypertension hos nyfödda
II. Pulmonell hypertension på basen av vänstersidig hjärtsjukdom
III. Pulmonell hypertension på basen av lungsjukdom och/eller hypoxi
IV. Kronisk tromboembolisk pulmonell hypertension
V. Pulmonell hypertension med oklara och/eller multifaktoriella mekanismer
Referenser
- Zarrinkoub R, Wettermark B, Wändell P, et al. The epidemiology of heart failure, based on data for 2.1 million inhabitants in Sweden. Eur J Heart Fail. 2013;15(9):995-1002.
- Hasenfuss G, Mann DL. Pathophysiology of heart failure. In: Mann DL, Zipes DL, Libby P, et al, editors. Braunwald’s Heart disease. 10th ed. Philadelphia: Saunders; 2015. p. 454-72.
- Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67-119.
- Vachiery JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100-8.
- Gerges C, Gerges M, Lang MB, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in »out-of-proportion« pulmonary hypertension. Chest. 2013;143(3):758-66.
- Naeije R, Vachiery JL, Yerly P, et al. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J. 2013;41(1):217-23.
- Enson Y, Wood JA, Mantaras NB, et al. The influence of heart rate on pulmonary arterial-left ventricular pressure relationships at end-diastole. Circulation. 1977;56(4 Pt 1):533-9.
- Gerges C, Gerges M, Lang IM. Characterization of pulmonary hypertension in heart failure using the diastolic pressure gradient: the conundrum of high and low diastolic pulmonary gradient. JACC Heart Fail. 2015;3(5):424-5.
- Naeije R. Measurement to predict survival: the case of diastolic pulmonary gradient. JACC Heart Fail. 2015;3(5):425.
- Tampakakis E, Tedford RJ. Reply: characterization of pulmonary hypertension in heart failure using the diastolic pressure gradient: the conundrum of high and low diastolic pulmonary gradient. JACC Heart Fail. 2015;3(5):426-7.
- Lundgren J, Rådegran G. Pathophysiology and potential treatments of pulmonaryhypertension due to systolic left heart failure. Acta Physiol (Oxf). 2014;211(2):314-33.
- Kurdak SS, Namba Y, Fu Z, et al. Effect of increased duration of high perfusion pressure on stress failure of pulmonary capillaries. Microvasc Res. 1995;50(2):235-48.
- West JB, Mathieu-Costello O. Vulnerability of pulmonary capillaries in heart disease. Circulation. 1995;92(3):622-31.
- Cooper CJ, Jevnikar FW, Walsh T, et al. The influence of basal nitric oxide activity on pulmonary vascular resistance in patients with congestive heart failure. Am J Cardiol. 1998;82(5):609-14.
- Ooi H, Colucci WS, Givertz MM. Endothelin mediates increased pulmonary vascular tone in patients with heart failure: demonstration by direct intrapulmonary infusion of sitaxsentan. Circulation. 2002;106(13):1618-21.
- Cody RJ, Haas GJ, Binkley PF, et al. Plasma endothelin correlates with the extent of pulmonary hypertension in patients with chronic congestive heart failure. Circulation. 1992;85(2):504-9.
- Tsutamoto T, Wada A, Maeda Y, et al. Relation between endothelin-1 spillover in the lungs and pulmonary vascular resistance in patients with chronic heart failure. J Am Coll Cardiol. 1994;23(6):1427-33.
- Fang JC, DeMarco T, Givertz MM, et al. World Health Organization Pulmonary Hypertension group 2: pulmonary hypertension due to left heart disease in the adult – a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2012;31(9):913-33.
- Delgado JF, Conde E, Sanchez V, et al. Pulmonary vascular remodeling in pulmonary hypertension due to chronic heart failure. Eur J Heart Fail. 2005;7(6):1011-6.
- Guha A, Amione-Guerra J, Park MH. Epidemiology of pulmonary hypertension in left heart disease. Prog Cardiovasc Dis. 2016;59(1):3-10.
- Gerges M, Gerges C, Pistritto AM, et al. Pulmonary hypertension in heart failure. Epidemiology, right ventricular function, and survival. Am J Respir Crit Care Med. 2015;192(10):1234-46.
- Miller WL, Grill DE, Borlaug BA. Clinical features, hemodynamics, and outcomes of pulmonary hypertension due to chronic heart failure with reduced ejection fraction: pulmonary hypertension and heart failure. JACC Heart Fail. 2013;1(4):290-9.
- Chang PP, Longenecker JC, Wang NY, et al. Mild vs severe pulmonary hypertension before heart transplantation: different effects on posttransplantation pulmonary hypertension and mortality. J Heart Lung Transplant. 2005;24(8):998-1007.
- Tampakakis E, Leary PJ, Selby VN, et al. The diastolic pulmonary gradient does not predict survival in patients with pulmonary hypertension due to left heart disease. JACC Heart Fail. 2015;3(1):9-16.
- Rosenkranz S, Gibbs JS, Wachter R, et al. Left ventricular heart failure and pulmonary hypertension. Eur Heart J. 2016;37(12):942-54.
- Lam CS, Roger VL, Rodeheffer RJ, et al. Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. J Am Coll Cardiol. 2009;53(13):1119-26.
- Ghio S, Gavazzi A, Campana C, et al. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol 2001;37(1):183-8.
- Opitz CF, Hoeper MM, Gibbs JS, et al. Pre-capillary, combined, and post-capillary pulmonary hypertension: a pathophysiological continuum. J Am Coll Cardiol. 2016;68(4):368-78.
- Califf RM, Adams KF, McKenna WJ, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: the Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1997;134(1):44-54.
- Gheorghiade M, Greene S, Butler J, et al; SOCRATES-REDUCED Investigators and Coordinators. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: the SOCRATES-REDUCED randomized trial. JAMA. 2015;314(21):2251-62.
- Lundgren J, Algotsson L, Kornhall B, et al. Preoperative pulmonary hypertension and its impact on survival after heart transplantation. Scand Cardiovasc J. 2014;48(1):47-58.
- Kutty RS, Parameshwar J, Lewis C, et al. Use of centrifugal left ventricular assist device as a bridge to candidacy in severe heart failure with secondary pulmonary hypertension. Eur J Cardiothorac Surg. 2013;43(6):1237-42.
Summary
Pulmonary hypertension (PH) is a serious complication to left heart disease (LHD), affecting a majority of the patients during the course of the disease. Initially, PH-LHD is caused by passive congestion of the pulmonary vessels due to increased left atrial pressures, a condition that is currently denoted as isolated post-capillary PH (Icp-PH). In the majority of patients the increased atrial pressure is a result of elevated left ventricular filling pressures. Furthermore, chronically elevated filling pressures may yield endothelial damage, resulting in structural and functional alterations in the pre-capillary bed with further elevation in pulmonary pressures as well as elevated vascular resistance, defined as combined precapillary and postcapillary PH (Cpc-PH). With previous definitions of PH-LHD it has been difficult to differentiate between the subgroups, so a new classification was presented in the 2015 PH guidelines. Despite PH-LHD being common and serious, specific therapies are lacking for the pulmonary component. Instead, treatments focus on optimizing the underlying cause of PH-LHD and involve medical as well as surgical therapies. In the present review we presents, based on the new guidelines, current knowledge on pathophysiological and pathobiological mechanisms, epidemiology, investigation and treatment of PH-LHD.