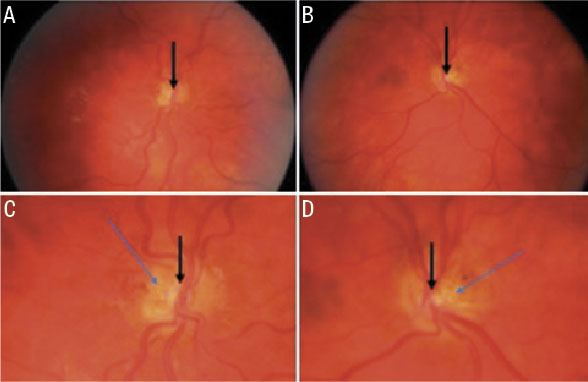
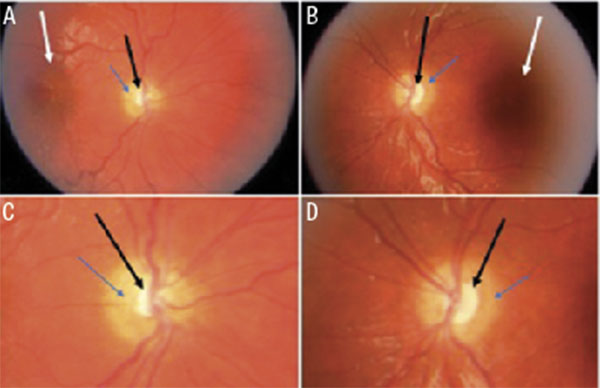


![Figur 6. A och B visar ögonbottenbilder från en 7 år gammal pojke med normal syn och normal papill på höger öga (A), men MGDA på vänster öga med en synskärpa på 0,05 (B). Bilder i figuren publicerade med tillstånd från Acta Ophthalmologica [83].](https://lakartidningen.se/wp-content/uploads/2020/10/fig6_w_.jpg)
Barn med medfödd optikushypoplasi, optikuskolobom eller MGDA (morning glory disc anomaly) debuterar ofta med dålig fixation, nystagmus, skelning och nedsatt syn men har hög risk för associerade sjukdomar, vilket kräver utredning.
Hypofysinsufficiens, neurologisk eller intellektuell funktionsnedsättning samt autism är överrepresenterade vid optikushypoplasi och optikuskolobom. Vid optikuskolobom förekommer även ökad risk för hjärtsjukdom och hörselnedsättning.
Encefalocele samt vaskulära, renala och endokrina problem kan finnas hos patienter med MGDA.
Optisk koherenstomografi kan ge värdefull hjälp vid klassificering och prognostisering av framtida synpotential.
β Förutom ögon- och barnläkarundersökning bör genetisk utredning övervägas.
Av världens barn under 15 års ålder är uppskattningsvis 19 miljoner synskadade, med en synskärpa lägre än 0,3. Av dessa är 1,4 miljoner barn blinda [1-3]. I låginkomstländer beror synskadan oftast på en grumlad hornhinna [4, 5] eller kongenital katarakt [6]. I hög- och medelinkomstländer har vaccinations- och screeningprogram samt tillgång till kataraktkirurgi ändrat synskadepanoramat [7]. Här är cerebral synskada, prematuritetsretinopati och optikushypoplasi viktiga orsaker till synskada [4, 8].
Hos 150 barn med kongenital eller infantil blindhet födda i Sverige 1988–2008 var orsakerna i fallande ordning: prematuritetsretinopati 24 procent, optikushypoplasi 15 procent och Lebers kongenitala amauros 15 procent. En majoritet av barnen hade ytterligare någon funktionsnedsättning [9].
Vi fokuserar i denna artikel på optikusmissbildningarna optikushypoplasi, optikuskolobom och MGDA (morning glory disc anomaly), som alla kan orsaka synskada och blindhet, med syftet att beskriva dessa ögonsjukdomar och även belysa dem ur ett barnmedicinskt samt genetiskt perspektiv på grund av den stora risken för associerade endokrinologiska och neurologiska sjukdomar.
Optikushypoplasi
Bakgrund och prevalens
Optikushypoplasi är en medfödd anläggningsstörning som innebär att synnerven är tunn och underutvecklad med färre gangliecellsaxoner än normalt. Optikushypoplasi var den vanligaste optikusmissbildningen i vår populationsbaserade Stockholmsstudie med en prevalens på 17/100 000 barn, varav cirka hälften av fallen (55 procent) var bilaterala [10]. Optikushypoplasi uppges i andra, möjligen selekterade, studier förekomma bilateralt i upp till 80 procent [11, 12].
Synnedsättningens spektrum spänner från subnormal synskärpa endast på ena ögat till bilateral total blindhet. Svår synskada/blindhet med en synskärpa på < 0,1 har uppgetts föreligga i 86 procent vid bilateral optikushypoplasi [13]. I Stockholmsstudien hade bara 36 procent av barnen med bilateral optikushypoplasi en synskärpa på < 0,1 [10]. Ett exempel på en uttalad ensidig optikushypoplasi men en normal papill i andra ögat ses i Figur 1.
Optikushypoplasi kan förekomma isolerat eller tillsammans med andra hjärnmissbildningar, neurologiska avvikelser eller hypofysinsufficiens [14-17].
Debutsymtom
Symtom vid svår bilateral optikushypoplasi hos det lilla barnet är till exempel att det inte reagerar på ljus, inte fixerar eller följer ansikten med blicken eller har nystagmus. Vid lindrigare eller ensidig optikushypoplasi upptäcks synnedsättningen ofta senare på grund av skelning, »titta-nära-beteende« eller vid rutinkontroll av synen på barnavårdscentralen eller i skolhälsovården. Mer sällan kan i stället de tidigare nämnda associerade sjukdomarna ge debutsymtom i form av neonatal recidiverande hypoglykemi, irritabilitet, kramp, ikterus, dålig tillväxt eller mikropenis.
Etiologi och riskfaktorer
Orsaken till det reducerade antalet gangliecellsaxoner vid optikushypoplasi kan vara tidig och primär, t ex bristande axonal vägledning, eller senare och sekundär till följd av ökad apoptos eller transsynaptisk degeneration [18-20]. Tidpunkten för skadan bestämmer skillnaden i storlek av papillen och exkavationen samt även graden av atrofi. En tidig skada under första trimestern resulterar ofta i en liten papill [19, 20].
Många etiologiska studier har visat att förstagångsgraviditet hos unga kvinnor är en oberoende riskfaktor för optikushypoplasi hos barnet [12, 21-23]. Vaginal blödning i tidig graviditet har rapporterats vara en riskfaktor [12], liksom exponering för olika teratogena substanser under embryogenesen [15]. Optikushypoplasi är beskrivet hos barn med fetalt alkoholsyndrom [24-26], men alkohol, liksom kongenitala infektioner, har i större kohortstudier visat sig vara en mer ovanlig orsak [23].
Genetiska orsaker är ännu inte fullständigt kartlagda. I de flesta fall verkar optikushypoplasi uppstå sporadiskt, men både punktmutationer och gendosavvikelser finns beskrivna, och nya sjukdomsorsakande gener upptäcks ständigt [27, 28].
Diagnostik
Diagnosen ställs kliniskt genom ögonbottenundersökning där papillstorleken bedöms i relation till andra ögats papill samt vidden på ögonbottenkärlen. Om möjligt kompletteras med ögonbottenfoto där man manuellt kan mäta papill–makula-avståndet och räkna ut en kvot [29]. Alternativt kan man beräkna papillarean digitalt [30].
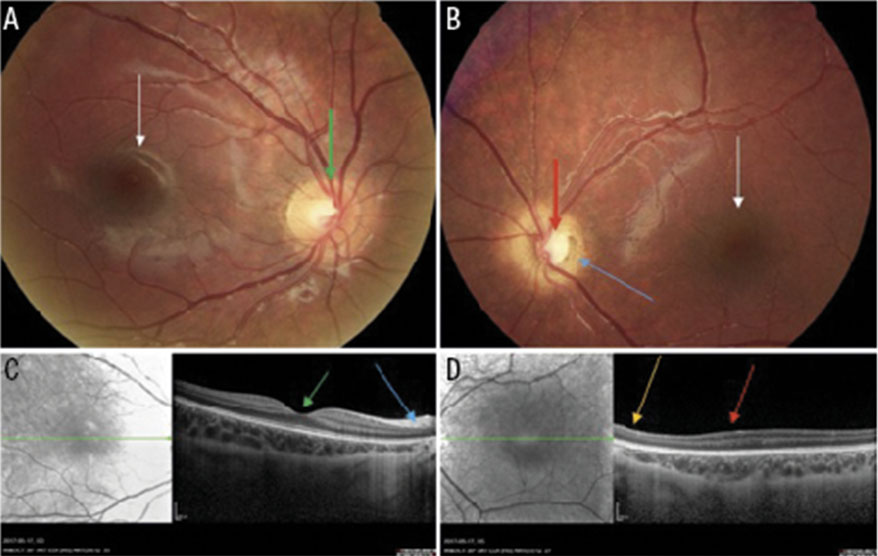
Förutom minskad storlek är papillen ofta blek och ibland omgiven av en dubbelring, vilken ses som en ljus halo, ibland omgiven av en pigmenterad kant [31] (Figur 1, 2 och 3). Dubbelringen kan göra att papillen ser falskt normalstor ut vid en snabb bedömning (Figur 1 och 3). Vallreflexen i makula kan saknas [10] (Figur 1 och 3), och arterioli och venoler kan vara slingriga eller abnormt raka [32, 33].
Optisk koherenstomografi (OCT), en skanning av näthinnans tjocklek med hjälp av laser, har visat sig ha hög sensitivitet och specificitet för diagnostik av optikushypoplasi. Förutom papillhypoplasi ses en förtunning av gangliecellslagret och nervfiberlagret (RNFL) samt en underutveckling av makula, s k foveal hypoplasi [34, 35] (Figur 1D). Förhoppningsvis kommer OCT i framtiden att förbättra möjligheten till säker diagnos samt synprognos, då synfunktion ofta är kopplad till foveal struktur och nervfiberlagret.
Med MR kan man vid optikushypoplasi ofta visualisera tunna synnerver och en avvikande chiasma [36] (Figur 4), men en normal neuroradiologisk undersökning utesluter inte optikushypoplasi och tunna synnerver kan också föreligga vid optikusatrofi [37].
Differentialdiagnoser
Optikusatrofi, förtvining av synnerven, utgör en differentialdiagnos [20]. Papillen är då normalstor, men blek och skarpt avgränsad. Synnedsättningen kommer ofta senare, men patienterna kan ha liknande OCT-förändringar som vid optikushypoplasi [38]. Optikusatrofi ses som ett resttillstånd efter hjärnskador som påverkat synnerven pre- eller postnatalt, såsom expansiv process eller hydrocefalus. Dominant optikusatrofi och Lebers hereditära optikusatrofi utgör genetiska orsaker, men där debuterar synnedsättningen ofta senare i barndomen eller i ungdomen.
Associerade tillstånd
Hypofysinsufficiens och strukturella avvikelser i CNS. Associationen mellan optikushypoplasi och hypofysinsufficiens beskrevs redan 1970 [14] och har verifierats i ett stort antal studier med prevalenssiffror varierande mellan 28 och 70 procent [11, 17, 39-45]. I den populationsbaserade Stockholmsstudien var prevalensen av hypofysinsufficiens 29 procent utan någon skillnad mellan den unilaterala och bilaterala gruppen [17]. Tidigare studier har där visat varierande resultat [42, 44, 46].
Tillväxthormonbrist är vanligast [41, 45, 47], följt av central hypotyreos [48] och brist på adrenokortikotropt hormon, som ger kortisolbrist vilket kan leda till hypoglykemi och hypotoni och vara livshotande vid allvarlig stress [49]. Dysfunktion av gonadotropiner kan ge pubertas praecox eller pubertas tarda, medan brist på antidiuretiskt hormon ger polyuri och polydipsi, det vill säga central diabetes insipidus. Hyperprolaktinemi är vanligt [39, 41, 45].
Vid optikushypoplasi och samtidig hypofysinsufficiens har det rapporterats en hög förekomst av neuroradiologiska strukturella avvikelser i adenohypofysen, infundibulum eller neurohypofysen (ektopisk hypofys eller avsaknad av neurohypofysen) (Figur 5) [17, 42, 44, 50]. Sådana avvikelser utgör viktiga prediktorer för hormonell insufficiens vid optikushypoplasi [42, 44, 50], men en strukturellt normal hypofys utesluter inte hypofysinsufficiens [15, 43, 45]. Även andra strukturella avvikelser i CNS, t ex septum pellucidum-agenesi, corpus callosum-hypoplasi/-agenesi och migrationsstörningar i hemisfärerna, förekommer [11, 17, 22, 41, 43, 51]. Avsaknad av septum pellucidum anses inte användbart som prognosmarkör för hypofysinsufficiens vid optikushypoplasi [17, 42, 43, 47]. Vidare är risken för strukturella avvikelser i CNS högre hos barn med bilateral optikushypoplasi [17, 43] och korrelerar till utvecklingsförseningar [15, 22, 43].
Neurologiska funktionsnedsättningar och autism. Redan på 1980-talet beskrevs barn med optikushypoplasi och samtidig intellektuell funktionsnedsättning, cerebral pares eller epilepsi [52]. Bilateral optikushypoplasi verkar öka risken för intellektuell funktionsnedsättning (prevalens 56 procent), motoriska svårigheter (74 procent), och epilepsi (23 procent) jämfört med unilateral optikushypoplasi, som dock också innebär en förhöjd risk [16, 17]. Även beteendeproblem är vanliga [10, 52-54], och autism har påvisats hos en tredjedel av blinda barn med optikushypoplasi [53, 55, 56]. I den populationsbaserade Stockholmsstudien, inkluderande barn med unilateral optikushypoplasi, var risken för autism 17 procent [16].
Utredning, behandling och prognos
Synnedsättningen vid optikushypoplasi är bestående, varför tidig remiss till syncentral vid misstanke om synskada är viktig för synhabilitering och optimering av synen för att minska negativ påverkan på övrig psykomotorisk utveckling. Barnet bör följas regelbundet hos ortoptist och ögonläkare i småbarnsåren och upp till 7–8 års ålder. Korrigering av större refraktionsfel med glasögon övervägs om det finns synpotential. Ocklusionsbehandling kan vara indicerad, men om svår optikushypoplasi föreligger bilateralt kan lappbehandling i stället försämra det bästa ögat och påverka barnets psykomotoriska utveckling negativt. När barnet är äldre kan skelningskirurgi övervägas för att bredda synfältet vid inåtskelning eller av psykosociala skäl. Nyttan av nystagmuskirurgi med bättre synskärpa och minskad nystagmus oberoende av bakomliggande orsak till synnedsättningen har beskrivits [57]. Efter åtta års ålder kan kontrollerna glesas ut om tillståndet är stabilt och om endokrinologiska och neurologiska komorbiditeter är väl kartlagda.
Det kan vara svårt att uttala sig om synprognos för det lilla barnet, men generellt kan sägas att synnedsättningen vid optikushypoplasi inte progredierar. I stället finns det en chans för viss mognad av synsystemet, tack vare ökad myelinisering av neuron i synbanorna, utveckling av synapser och mognad av primära och sekundära syncentra i hjärnan [58, 59]. Denna mognad verkar ske senare hos barn med optikushypoplasi [60], varför spädbarnet med optikushypoplasi som initialt förefaller vara helt blint ändå kan utveckla någon typ av synförmåga.
Vid upptäckt av såväl ensidig som dubbelsidig optikushypoplasi är skyndsam remittering till barnendokrinolog och barnneurolog obligat på grund av de tidigare beskrivna associerade tillstånden. Den pediatriska bedömningen inkluderar endokrinologisk utredning samt eventuellt MR av hjärna, orbita och hypofys. Barnens utveckling ska bedömas, och vid tecken till neuropsykiatrisk funktionsnedsättning ska vidare utredning initieras [16]. Barnen bör följas över tid, då till exempel hypofysinsufficiens kan utvecklas senare [15, 17] och autismdiagnos inte säkert kan ställas i låg ålder [55]. Genetisk utredning med så kallad genpanel, vilket innebär att ett urval av gener som är relevanta för den specifika symtombilden hos patienten analyseras, bör övervägas. I vissa fall kan även gendosarray och helgenomsekvensering vara motiverad.
Optikuskolobom
Bakgrund och prevalens
Ordet »kolobom« kommer av grekiskan och betyder stympad. Kolobom beror på en ofullständig slutning av fissuren i ögat under embryogenesen, vilket kan leda till defekter i såväl iris (nyckelhålspupill) som lins och ögonbotten, det senare i form av optikuskolobom med eller utan retinokoroidalt kolobom. Optikuskolobom är den näst vanligaste förekommande optikusmissbildningen bland Stockholmsbarn med en prevalens på 9/100 000, lika ofta ensidig som dubbelsidig [61].
Debutsymtom
De vanligaste tecknen är synnedsättning, skelning och/eller nystagmus. Även yttre förändringar som mikroftalmus och iriskolobom kan föreligga, och vid ett samtidigt stort retinokoroidalt kolobom kan pupillen vara vit. Graden av synnedsättning vid optikuskolobom varierar och beror bland annat på kolobomets storlek och lokalisation [62].
Etiologi och riskfaktorer
Uppkomstmekanismen är en slutningsdefekt av de inre lagren av den fetala optikusstjälken i graviditetsvecka 5–7 [63-65]. Därefter kan inte heller de yttre lagren sluta sig och ger i stället upphov till ett gap, hål eller fissur, och i vissa fall en cysta. Slutningsdefekten inhiberar troligtvis normal tillväxt av hela ögonblåsan, vilket gör att kolobom, framför allt optikuskolobom, är associerat med mikroftalmus [63, 65, 66].
Majoriteten av optikuskolobom-fallen är sporadiska och av oklar etiologi. Autosomalt dominant nedärvning är vanligast vid isolerat optikuskolobom, men även andra nedärvningsmönster finns beskrivna [64, 66]. Mutationer i generna PAX2, PAX6, SOX2, OTX2 och RX är vanliga [64, 65, 67]. Även teratogena substanser som talidomid, LSD, antiepileptika, alkohol och kokain har kopplats till optikuskolobom [63, 66].
Diagnostik
Diagnosen optikuskolobom är klinisk och ställs genom ögonbottenundersökning. Omfattningen av optikuskolobom kan variera från en förstorad papillexkavation till ett utbrett papilloretinokoroidalt kolobom som engagerar också omgivande retina och koroidea (Figur 5B). Då kan fotografering med vidvinkelkamera vara av värde. OCT kan påvisa sekundärkomplikationer som subretinal vätska och näthinneavlossning [68].
Differentialdiagnoser
Tillstånd som man kan förväxla med optikuskolobom är makulakolobom, »optic pit«, peripapillärt stafylom och MGDA [69, 70].
Associerade tillstånd och komplikationer. Kolobom kan vara en isolerad förändring hos ett i övrigt friskt barn. Dock förekommer hörselnedsättning, beteendeförändringar, hypofysinsufficiens samt intellektuell och neurologisk funktionsnedsättning i ökad grad hos barn med optikuskolobom [61, 65, 71]. Ibland är kolobomet en del av ett syndrom, såsom CHARGE-, Kabuki- [72, 73], Joubert- [71, 74] eller cat eye-syndromet [75].
CHARGE ingår i gruppen dövblindsyndrom och orsakas av en mutation i CHD7-genen. Namnet är en akronym av de engelska orden för kolobom, hjärtfel, koanalatresi, tillväxthämning, underutveckling av könsorganen och öronmissbildning/hörselnedsättning/dövhet. Barn med CHARGE drabbas av flera eller alla av ovan nämnda tillstånd i varierande svårighetsgrad [76]. Kolobom förekommer hos 86 procent av barnen, men även mikroftalmus och optikushypoplasi kan förekomma [77]. Risken för näthinneavlossning är ökad hos barn med optikuskolobom [61, 78-81] framför allt vid samtidigt retinokoroidalt kolobom. Risken för amblyopi är också ökad, speciellt vid samtidig mikroftalmus och anisometropi [63].
Utredning, behandling och prognos
Synprognosen varierar och beror på omfattning och utbredning av optikuskolobom [68], där makulaengagemang och mikroftalmus är ogynnsamma faktorer [61, 63]. Precis som vid optikushypoplasi sträcker sig spektrumet från god synskärpa utan associerade tillstånd till blindhet bilateralt och associerade intellektuella och neurologiska funktionsnedsättningar [61, 63]. Ingen progressiv synnedsättning har dock dokumenterats om inte komplikationer, såsom t ex näthinne-
avlossning eller amblyopi, har uppstått [70]. Det är därför av stor vikt att följa barnen regelbundet under småbarnsåren för att tidigt upptäcka och behandla detta. Efter 7–8 års ålder kan kontrollerna glesas ut.
Remiss till barnläkare bör alltid övervägas då förekomst av kolobom kan motivera ytterligare utredning hos det multisjuka barnet [61]. Beroende på vilken diagnos som misstänks görs olika genetiska analyser, exempelvis genpanel eller gendosarray. Det är också viktigt med hörselkontroller.
MGDA (Morning Glory Disc Anomaly)
Bakgrund och prevalens
MGDA är oftast en ensidig optikusmissbildning där man har en trattliknande utbuktning av bakre skleraväggen inkluderande synnerven. Utseendet påminner om blomman »morning glory« (»blomman för dagen« på svenska) [82]. Orsaken tros bero på dålig utveckling av mesenkymet i bakre sklera och lamina cribrosa under fosterstadiet [70]. Förekomsten i en populationsbaserad studie i Stockholm var 2,6/100 000 [83].
Debutsymtom
MGDA debuterar ofta med skelning, och när barnet är äldre kan en relativt uttalad synnedsättning påvisas [84, 85].
Diagnostik
Diagnosen MGDA är klinisk och ställs genom ögonbottenspegling, där man ser en förstorad papill med peripapillära pigmenteringar, en djup exkavation, ett radierande mönster av retinala kärl och en vit gliatofs ovan papillen [70] (Figur 6B). Risk för serös näthinneavlossning föreligger [83, 85]. Man kan också se typiska tecken på MR som trattformad exkavation och avsaknad av sklera baktill på bulben [86].
Differentialdiagnoser
Till differentialdiagnoserna räknas optikuskolobom, optikusgliom och bakre stafylom [82].
Associerade tillstånd. Neurologiska, neurovaskulära, kardiovaskulära, renala och endokrina problem kan finnas hos patienter med MGDA. Även medellinjedefekter med brett avstånd mellan ögonen, läpp-, käk- och gomspalt och cerebrala abnormiteter som basalt encefalocele, cerebrovaskulära anomalier, moyamoyasjukdom, hypofyspåverkan och corpus callosum-agenesi förekommer varför MR av hjärnan rekommenderas [83, 84, 87-89].
Utredning, behandling och prognos
Amblyopibehanding är viktig att initiera vid MGDA [90], och skelningskirurgi kan bli aktuell. Serös retinal avlossning förekommer i ca 30 procent [83] och kräver oftast bara uppföljning, men glaskroppskirurgi med laserbehandling kan bli aktuell. MR av orbita och hjärnan visar breddökningen av synnerven som kan likna optikusgliom, och på MR-angiografi ses också basala encefalocelen och moyamoyasjuka [91].
Sammanfattning
Barn med olika typer av optikusmissbildningar diagnostiseras och följs i huvudsak på ögonklinik och eventuellt på syncentral. Vid såväl ensidig som dubbelsidig optikushypoplasi, optikuskolobom eller MGDA finns det dock risk för komorbiditet. Kunskap om risken för associerade problem bör återfinnas multidisciplinärt hos barnoftalmologer, ortoptister, barnneurologer, barnendokrinologer, ÖNH-läkare, audiologer, barnpsykiatrer, pedagoger och syncentralsoptiker. Detta för att kunna sätta tidig diagnos, initiera adekvat utredning, behandling och habilitering; allt för att optimera barnens förutsättningar och livskvalitet.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Referenser
- Resnikoff S, Pascolini D, Etya’ale D, et al. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82(11):844-51.
- Pascolini D, Mariotti SP. Global estimates of visual impairment: 2010. Br J Ophthalmol. 2012;96(5):614-8.
- World Health Organization (WHO). ICD-11 for mortality and morbidity statistics. 9D90 Vision impairment including blindness. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/1103667651
- Kong L, Fry M, Al-Samarraie M, et al. An update on progress and the changing epidemiology of causes of childhood blindness worldwide. J AAPOS. 2012;16(6):501-7.
- Semba RD, Bloem MW. Measles blindness. Surv Ophthalmol. 2004;49(2):243-55.
- Maida JM, Mathers K, Alley CL. Pediatric ophthalmology in the developing world. Curr Opin Ophthalmol. 2008;19(5):403-8.
- Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-7.
- Blohmé J, Tornqvist K. Visual impairment in Swedish children. III. Diagnoses. Acta Ophthalmol Scand. 1997;75(6):681-7.
- de Verdier K, Ulla E, Löfgren S, et al. Children with blindness – major causes, developmental outcomes and implications for habilitation and educational support: a two-decade, Swedish population-based study. Acta Ophthalmol. 2018;96(3):295-300.
- Teär Fahnehjelm K, Dahl S, Martin L, et al. Optic nerve hypoplasia in children and adolescents; prevalence, ocular characteristics and behavioural problems. Acta Ophthalmol. 2014;92(6):563-70.
- Skarf B, Hoyt CS. Optic nerve hypoplasia in children. Association with anomalies of the endocrine and CNS. Arch Ophthalmol. 1984;102(1):62-7.
- Garcia-Filion P, Fink C, Geffner ME, et al. Optic nerve hypoplasia in North America: a re-appraisal of perinatal risk factors. Acta Ophthalmol. 2010;88(5):527-34.
- Siatkowski RM, Sanchez JC, Andrade R, et al. The clinical, neuroradiographic, and endocrinologic profile of patients with bilateral optic nerve hypoplasia. Ophthalmology. 1997;104(3):493-6.
- Hoyt WF, Kaplan SL, Grumbach MM, et al. Septo-optic dysplasia and pituitary dwarfism. Lancet. 1970;1(7652):893-4.
- Ryabets-Lienhard A, Stewart C, Borchert M, et al. The optic nerve hypoplasia spectrum: review of the literature and clinical guidelines. Adv Pediatr. 2016;63(1):127-46.
- Dahl S, Wickström R, Ek U, et al. Children with optic nerve hypoplasia face a high risk of neurodevelopmental disorders. Acta Paediatr. 2018;107(3):484-9.
- Dahl S, Kristoffersen Wiberg M, Teär Fahnehjelm K, et al. High prevalence of pituitary hormone deficiency in both unilateral and bilateral optic nerve hypoplasia. Acta Paediatr. 2019;108(9):1677-85.
- Frisen L, Holmegaard L. Spectrum of optic nerve hypoplasia. Br J Ophthalmol. 1978;62(1):7-15.
- Jacobson L, Hellström A, Flodmark O. Large cups in normal-sized optic discs: a variant of optic nerve hypoplasia in children with periventricular leukomalacia. Arch Ophthalmol. 1997;115(10):1263-9.
- Hoyt CS, Good WV. Do we really understand the difference between optic nerve hypoplasia and atrophy? Eye (Lond). 1992;6(Pt 2):201-4.
- Tornqvist K, Ericsson A, Kallen B. Optic nerve hypoplasia: risk factors and epidemiology. Acta Ophthalmol Scand. 2002;80(3):300-4.
- Goh YW, Andrew D, McGhee C, et al. Clinical and demographic associations with optic nerve hypoplasia in New Zealand. Br J Ophthalmol. 2014;98(10):1364-7.
- Garcia-Filion P, Borchert M. Prenatal determinants of optic nerve hypoplasia: review of suggested correlates and future focus. Surv Ophthalmol. 2013;58(6):610-9.
- Strömland K. Ocular abnormalities in the fetal alcohol syndrome. Acta Ophthalmol Suppl. 1985;171:1-50.
- Strömland K. Visual impairment and ocular abnormalities in children with fetal alcohol syndrome. Addict Biol. 2004;9(2):153-7; discussion 159-60.
- Ribeiro IM, Vale PJ, Tenedorio PA, et al. Ocular manifestations in fetal alcohol syndrome. Eur J Ophthalmol. 2007;17(1):104-9.
- Chen CA, Yin J, Lewis RA, et al. Genetic causes of optic nerve hypoplasia. J Med Genet. 2017;54(7):441-9.
- Dahl S, Pettersson M, Eisfeldt J, et al. Whole genome sequencing unveils genetic heterogeneity in optic nerve hypoplasia. PLoS One. 2020;15(2):e0228622.
- Zeki SM, Dudgeon J, Dutton GN. Reappraisal of the ratio of disc to macula/disc diameter in optic nerve hypoplasia. Br J Ophthalmol. 1991;75(9):538-41.
- Bartling H, Wanger P, Martin L. Measurement of optic disc parameters on digital fundus photographs: algorithm development and evaluation. Acta Ophthalmol. 2008;86(8):837-41.
- Mosier MA, Lieberman MF, Green WR, et al. Hypoplasia of the optic nerve. Arch Ophthalmol. 1978;96(8):1437-42.
- Hellström A, Wiklund LM, Svensson E, et al. Optic nerve hypoplasia with isolated tortuosity of the retinal veins: a marker of endocrinopathy. Arch Ophthalmol. 1999;117(7):880-4.
- Garcia-Filion P, Borchert M. Optic nerve hypoplasia syndrome: a review of the epidemiology and clinical associations. Curr Treat Opt Neurol. 2013;15(1):78-89.
- Pilat A, Sibley D, McLean RJ, et al. High-resolution imaging of the optic nerve and retina in optic nerve hypoplasia. Ophthalmology. 2015;122(7):1330-9.
- Katagiri S, Nishina S, Yokoi T, et al. Retinal structure and function in eyes with optic nerve hypoplasia. Sci Rep. 2017;7:42480.
- Brodsky MC, Glasier CM, Pollock SC, et al. Optic nerve hypoplasia. Identification by magnetic resonance imaging. Arch Ophthalmol. 1990;108(11):1562-7.
- Parravano JG, Toledo A, Kucharczyk W. Dimensions of the optic nerves, chiasm, and tracts: MR quantitative comparison between patients with optic atrophy and normals. J Comput Assist Tomogr. 1993;17(5):688-90.
- Barboni P, Savini G, Carelli V, et al. Re: Pilat et al: High-resolution imaging of the optic nerve and retina in optic nerve hypoplasia (Ophthalmology. 2015;122:1330-9). Ophthalmology. 2016;123(3):e19-20.
- Margalith D, Tze WJ, Jan JE. Congenital optic nerve hypoplasia with hypothalamic-pituitary dysplasia. A review of 16 cases. Am J Dis Child. 1985;139(4):361-6.
- Birkebaek NH, Patel L, Wright NB, et al. Endocrine status in patients with optic nerve hypoplasia: relationship to midline central nervous system abnormalities and appearance of the hypothalamic-pituitary axis on magnetic resonance imaging. J Clin Endocrinol Metab. 2003;88(11):5281-6.
- Garcia-Filion P, Epport K, Nelson M, et al. Neuroradiographic, endocrinologic, and ophthalmic correlates of adverse developmental outcomes in children with optic nerve hypoplasia: a prospective study. Pediatrics. 2008;121(3):e653-9.
- Ramakrishnaiah RH, Shelton JB, Glasier CM, et al. Reliability of magnetic resonance imaging for the detection of hypopituitarism in children with optic nerve hypoplasia. Ophthalmology. 2014;121(1):387-91.
- Garcia-Filion P, Almarzouki H, Fink C, et al. Brain malformations do not predict hypopituitarism in young children with optic nerve hypoplasia. Horm Res Paediatr. 2017;88(3-4):251-7.
- Alyahyawi N, Dheensaw K, Islam N, et al. Pituitary dysfunction in pediatric patients with optic nerve hypoplasia: a retrospective cohort study (1975-2014). Horm Res Paediatr. 2018;89(1):22-30.
- Oatman OJ, McClellan DR, Olson ML, et al. Endocrine and pubertal disturbances in optic nerve hypoplasia, from infancy to adolescence. Int J Pediatr Endocrinol. 2015;2015(1):8.
- Ahmad T, Garcia-Filion P, Borchert M, et al. Endocrinological and auxological abnormalities in young children with optic nerve hypoplasia: a prospective study. J Pediatr. 2006;148(1):78-84.
- Ahmad T, Borchert M, Geffner M. Optic nerve hypoplasia and hypopituitarism. Pediatr Endocrinol Rev. 2008;5(3):772-7.
- Ma NS, Fink C, Geffner ME, et al. Evolving central hypothyroidism in children with optic nerve hypoplasia. J Pediatr Endocrinol Metab. 2010;23(1–2):53-8.
- Brodsky MC, Conte FA, Taylor D, et al. Sudden death in septo-optic dysplasia. Report of 5 cases. Arch Ophthalmol. 1997;115(1):66-70.
- Phillips PH, Spear C, Brodsky MC. Magnetic resonance diagnosis of congenital hypopituitarism in children with optic nerve hypoplasia. J AAPOS. 2001;5(5):275-80.
- Zeki SM, Hollman AS, Dutton GN. Neuroradiological features of patients with optic nerve hypoplasia. J Pediatr Ophthalmol Strabismus. 1992;29(2):107-12.
- Margalith D, Jan JE, McCormick AQ, et al. Clinical spectrum of congenital optic nerve hypoplasia: review of 51 patients. Dev Med Child Neurol. 1984;26(3):311-22.
- Teär Fahnehjelm K, Wide K, Flodmark O, et al. Posterior ocular malformations in children: somatic, neuroradiological and cognitive aspects. Acta Paediatr. 2003;92(3):301-8.
- Ek U, Fernell E, Jacobson L. Cognitive and behavioural characteristics in blind children with bilateral optic nerve hypoplasia. Acta Paediatr. 2005;94(10):1421-6.
- Parr JR, Dale NJ, Shaffer LM, et al. Social communication difficulties and autism spectrum disorder in young children with optic nerve hypoplasia and/or septo-optic dysplasia. Dev Med Child Neurol. 2010;52(10):917-21.
- Jutley-Neilson J, Harris G, Kirk J. The identification and measurement of autistic features in children with septo-optic dysplasia, optic nerve hypoplasia and isolated hypopituitarism. Res Dev Disabil. 2013;34(12):4310-8.
- Dubner M, Nelson LB, Gunton KB, et al. Clinical evaluation of four-muscle tenotomy surgery for nystagmus. J Pediatr Ophthalmol Strabismus. 2016;53(1):16-21.
- Atkinson J. Human visual development over the first 6 months of life. A review and a hypothesis. Hum Neurobiol. 1984;3(2):61-74.
- Tabuchi A. Dynamic topography of visual evoked potential in children: a study of the development of the visual system. Jpn J Ophthalmol. 1985;29(2):153-60.
- Fielder AR, Fulton AB, Mayer DL. Visual development of infants with severe ocular disorders. Ophthalmology. 1991;98(8):1306-9.
- Skriapa Manta A, Olsson M, Ek U, et al. Optic disc coloboma in children – prevalence, clinical characteristics and associated morbidity. Acta Ophthalmol. 2019;97(5):478-85.
- Gopal L, Badrinath SS, Kumar KS, et al. Optic disc in fundus coloboma. Ophthalmology. 1996;103(12):2120-6; discussion 2126-7.
- Pagon RA. Ocular coloboma. Surv Ophthalmol. 1981;25(4):223-36.
- Patel A, Sowden JC. Genes and pathways in optic fissure closure. Semin Cell Dev Biol. 2019;91:55-65.
- Ohuchi H, Sato K, Habuta M, et al. Congenital eye anomalies: more mosaic than thought? Congenit Anom (Kyoto). 2019;59(3):56-73.
- ALSomiry AS, Gregory-Evans CY, Gregory-Evans K. An update on the genetics of ocular coloboma. Hum Genet. 2019;138(8–9):865-80.
- Slavotinek A. Genetics of anophthalmia and microphthalmia. Part 2: Syndromes associated with anophthalmia-microphthalmia. Hum Genet. 2019;138(8–9):831-46.
- Gopal L. A clinical and optical coherence tomography study of choroidal colobomas. Curr Opin Ophthalmol. 2008;19(3):248-54.
- Lee KM, Woo SJ, Hwang JM. Evaluation of congenital excavated optic disc anomalies with spectral-domain and swept-source optical coherence tomography. Graefes Arch Clin Exp Ophthalmol. 2014;252(11):1853-60.
- Jeng-Miller KW, Cestari DM, Gaier ED. Congenital anomalies of the optic disc: insights from optical coherence tomography imaging. Curr Opin Ophthalmol. 2017;28(6):579-86.
- Dijk DR, Bocca G, van Ravenswaaij-Arts CM. Growth in CHARGE syndrome: optimizing care with a multidisciplinary approach. J Multidiscip Healthc. 2019;12:607-20.
- Verhagen JM, Oostdijk W, Terwisscha van Scheltinga CE, et al. An unusual presentation of Kabuki syndrome: clinical overlap with CHARGE syndrome. Eur J Med Genet. 2014;57(9):510-2.
- Patel N, Alkuraya FS. Overlap between CHARGE and Kabuki syndromes: more than an interesting clinical observation? Am J Med Genet A. 2015;167A(1):259-60.
- Wang SF, Kowal TJ, Ning K, et al. Review of ocular manifestations of Joubert syndrome. Genes (Basel). 2018;9(12):605.
- Zellweger H, Ionasescu V, Simpson J et al. The problem of trisomy 22. A case report and a discussion of the variant forms. Clin Pediatr (Phila). 1976;15(7):601-18.
- Hsu P, Ma A, Wilson M, et al. CHARGE syndrome: a review. J Paediatr Child Health. 2014;50(7):504-11.
- Russell-Eggitt IM, Blake KD, Taylor DS, et al. The eye in the CHARGE association. Br J Ophthalmol. 1990;74(7):421-6.
- Jesberg DO, Schepens CL. Retinal detachment associated with coloboma of the choroid. Arch Ophthalmol. 1961;65:163-73.
- Daufenbach DR, Ruttum MS, Pulido JS, et al. Chorioretinal colobomas in a pediatric population. Ophthalmology. 1998;105(8):1455-8.
- Hussain RM, Abbey AM, Shah AR, et al. Chorioretinal coloboma complications: retinal detachment and choroidal neovascular membrane. J Ophthalmic Vis Res. 2017;12(1):3-10.
- Chen YN, Patel CK, Kertes PJ, et al. Retinal detachment and retrobulbar cysts in a large cohort of optic nerve coloboma. Retina. 2018;38(4):692-7.
- Lee BJ, Traboulsi EI. Update on the morning glory disc anomaly. Ophthalm Genet. 2008;29(2):47-52.
- Ceynowa DJ, Wickström R, Olsson M, et al. Morning glory disc anomaly in childhood – a population-based study. Acta Ophthalmol. 2015;93(7):626-34.
- Chen CS, David D, Hanieh A. Morning glory syndrome and basal encephalocele. Childs Nerv Syst. 2004;20(2):87-90.
- Chang S, Gregory-Roberts E, Chen R. Retinal detachment associated with optic disc colobomas and morning glory syndrome. Eye (Lond). 2012;26(4):494-500.
- Ellika S, Robson CD, Heidary G, et al. Morning glory disc anomaly: characteristic MR imaging findings. AJNR Am J Neuroradiol. 2013;34(10):2010-4.
- Caprioli J, Lesser RL. Basal encephalocele and morning glory syndrome. Br J Ophthalmol. 1983;67(6):349-51.
- Rao RC, Larson SA, Dlouhy BJ. Growth hormone deficiency, basal encephalocele, and morning glory anomaly. Ophthalmology. 2017;124(10):1531.
- Loddenkemper T, Friedman NR, Ruggieri PM, et al. Pituitary stalk duplication in association with moya moya disease and bilateral morning glory disc anomaly – broadening the clinical spectrum of midline defects. J Neurol. 2008;255(6):885-90.
- Cavazos-Adame H, Olvera-Barrios A, Martinez-Lopez-Portillo A, et al. Morning glory disc anomaly, a report of a successfully treated case of functional amblyopia. J Clin Diagn Res. 2015;9(10):ND01-3.
- Thoma D, Nijs I, Demaerel P, et al. Morning glory disc anomaly with an ipsilateral enlargement of the optic nerve pathway. Eur J Paediatr Neurol. 2017;21(5):787-91.
Summary
Congenital optic disc malformations can cause visual impairment. Diagnostics can be challenging during early infancy and childhood and visual prognosis difficult to predict; optical coherence tomography may be of value. The children often have coexisting comorbidities like pituitary hormone deficiency, neurodevelopmental disorders, and neurological impairments, and evaluation by a pediatrician should be performed. Also, genetic analyses should be considered. Co-operation in multidisciplinary teams is of value for correct diagnosis, visual habilitation and treatment of hormonal and neurological dysfunctions.