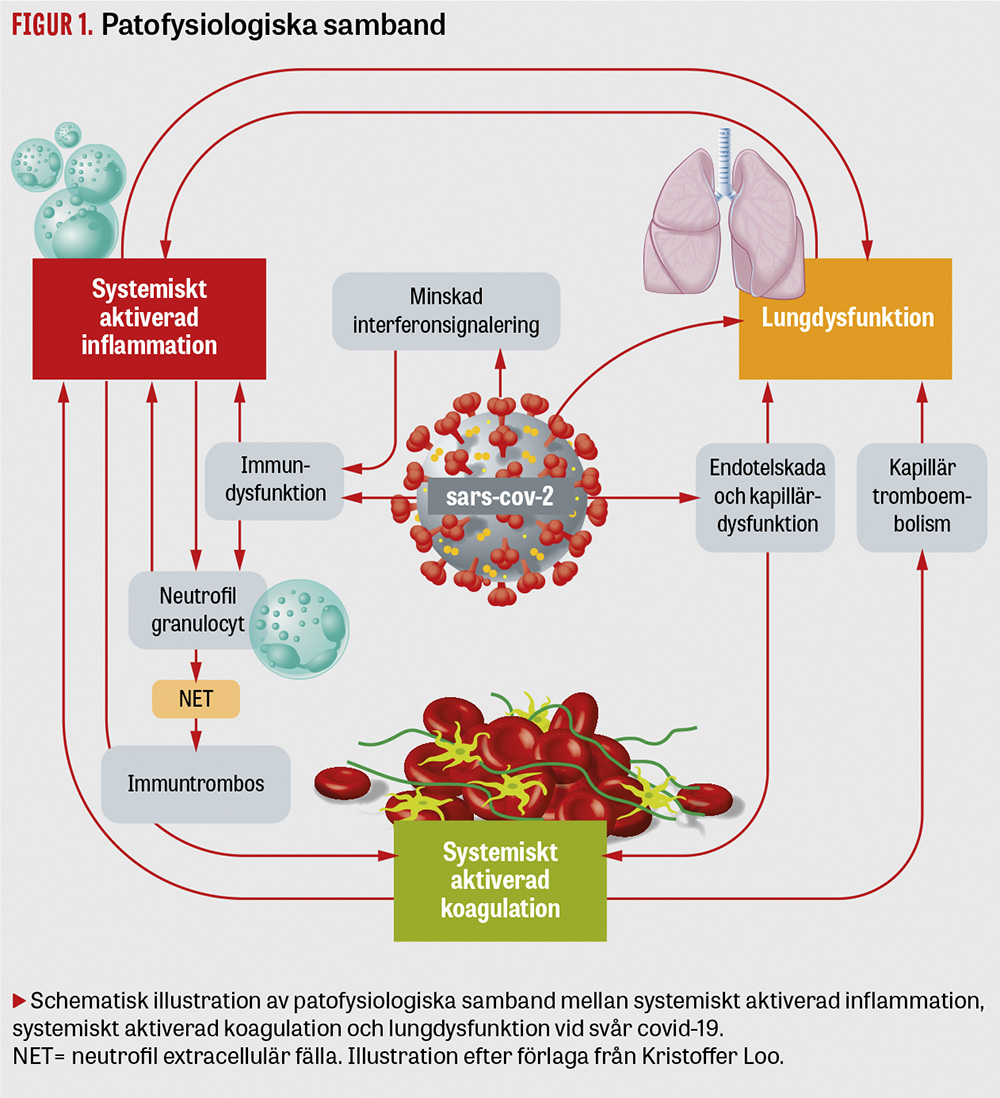
Patofysiologin bakom svår covid-19 drivs mer av aktiverad inflammation och koagulation än av direkta viruseffekter.
Ett dåligt reglerat cellulärt och molekylärt immunsvar vid svår covid-19 utlöser en självförstärkande spiral av okontrollerad systemisk inflammation och koagulopati, som i första hand skadar alveoler och kapillärendotel i lungorna.
Endotelskador i lungorna vid ARDS utlöst av covid-19 är vanligare och mer utbredda än vid ARDS av annan infektiös orsak.
Dödligheten vid svår covid-19 är hög.
Mikrotrombotisering bedöms vara den viktigaste orsaken till död i covid-19.
Störst nytta av immunmodulerande behandling vid svår covid-19 kan förväntas vid samtidigt avtagande virusinfektion och tilltagande inflammation.
Covid-19 är beteckningen på den sjukdom som främst drabbar lungorna efter att man blivit smittad med sars-cov-2. Feber, andfåddhet, hosta, muskelvärk och allmän sjukdomskänsla är vanliga debutsymtom, och ofta dröjer det 7–10 dagar innan den smittade utvecklar svår andningspåverkan med hypoxi som kräver sjukhusvård [1]. Dödsfall i covid-19 föregås ofta av akut lungsvikt, septisk chock och multipel organsvikt [2]. Tidigt under pandemin noterades en hög incidens av tromboemboliska komplikationer vid covid-19. I en metaanalys innefattande mer än 11 000 sjukhusvårdade patienter i 36 studier var incidensen av venös tromboembolism 17 procent [3]. I obduktionsmaterial förelåg trombotisering av såväl små som stora lungkärl hos avlidna patienter med covid-19 [4]. Kombinationen grav hypoxi med relativt väl bevarad lungcompliance fick läkare att överväga om lungsvikten i covid-19 är beroende av trombotisering av lungkärl [5].
I denna översikt lyfter vi fram några patofysiologiska mekanismer som rapporterats vara involverade i utvecklingen av svår covid-19 samt deras interaktioner (Figur 1).
Cytokiner
Sars-cov-2 inducerar vid svår covid-19 ett obalanserat immunförsvar som i förlängningen bidrar till morbiditet och mortalitet. Vid svår covid-19 noteras höga uttryck av nuclear factor (NF)-κB-beroende pro-inflammatoriska cytokiner som interleukin-6 (IL-6) och tumörnekrosfaktor alfa (TNF-α), medan uttryck av typ-1-interferoner (IFN), som är viktiga delar av det initiala immunförsvaret vid virusinfektion, rapporterats vara obefintliga (IFN-β) eller mycket låga (IFN-α) [6]. Förhöjda nivåer av IL-6 vid ankomst till sjukhus med covid-19 har rapporterats vara en starkare negativ prognostisk faktor än förhöjda inflammationsmarkörer som CRP och ferritin eller förhöjd D-dimer i en stor amerikansk studie [7], även om nivåerna av IL-6 inte är fullt så höga vid covid-19 som vid många andra tillstånd med hyperinflammation och akut svår lungsvikt (ARDS) [8]. Vid virusorsakad vävnadsskada i lungorna frisätter makrofager och endotelceller IL-8, som kemotaktiskt rekryterar neutrofila granulocyter (se vidare nedan). Genom kraftig frisättning av IL-6 startar dessa granulocyter, under inverkan av cytokiner samt aktivering av toll-liknande receptor 8 (TLR-8) via extracellulär ribonukleinsyra (RNA), en självförstärkande negativ spiral av inflammation och vävnadsskada [9-11].
Immunceller
Lungornas stora alveolaryta, som kontinuerligt exponeras för mikroorganismer från omgivningen, har ett välutvecklat och effektivt lokalt immunförsvar. I lungcirkulationen återfinns uppskattningsvis 40 procent av kroppens neutrofila granulocyter [12], som bedöms ha en central patofysiologisk roll för uppkomst av svår covid-19. Vid ARDS, som kan vara en del i svår covid-19, återfinns neutrofiler i alveolarvätska [13, 14]. Neutrofila granulocyter är potenta immunförsvarsceller som kan bekämpa invaderande patogener med bland annat neutrofila extracellulära fällor (NET) och reaktiva syreföreningar (ROS) [13]. Förhöjda nivåer av NET, som utgörs av kromatin och proteaser, föreligger ofta i blod, trakealsekret och lungvävnad vid svår covid-19 [9, 15]. Molekylära aktiveringsvägar för insöndring av NET är ofullständigt kända, men invaderande patogener, proinflammatoriska cytokiner samt aktiverade trombocyter och immunkomplex har föreslagits kunna vara involverade [16]. Neutrofila granulocyter från friska forskningspersoner frisätter NET vid direkt kontakt med sars-cov-2 in vitro [15]. Olika samband mellan NET och vävnadsskada vid covid-19 har föreslagits, och NET har rapporterats framkalla såväl alveolär apoptos in vitro [15] som trombocytaktivering med intravaskulär koagulation och kapillär mikrotrombotisering i enlighet med konceptet immuntrombos [17]. Samband har vid covid-19 rapporterats mellan antalet NET i plasma, behov av intubation och död [18]. Därmed kan NET utgöra en möjlig länk i det komplexa samspelet mellan viruspartiklar, cellulärt och molekylärt immunförsvar samt koagulation. Vidare uppvisar patienter med svår covid-19 ofta en patologisk blodbild med neutrofili i kombination med lymfopeni, där en högre granulocyt/lymfocytkvot avspeglar en svårare sjukdomsbild med högre risk för ARDS och död [9, 19-21].
Dessutom har lungskador hos personer avlidna med covid-19 visats vara associerade med perivaskulär infiltration av T-lymfocyter [22]. Det T-cellsmedierade immunförsvaret verkar vid svår covid-19 vara nedsatt med lymfopeni i perifert blod och T-cellsdysfunktion, vilket bidrar till försämrad kontroll av infektionen och spär på immunmedierade lungskador [23].
Även andra rubbningar av det cellulära immunförsvaret har beskrivits som konsekvenser av covid-19 eller som bidragande faktorer till patogenesen vid svår covid-19. Patienter med svår covid-19 uppvisar högre grad av aktivering och genetiskt uttryck av naturliga mördarceller (NK-celler) [24], stora granulära lymfocyter i det icke-adaptiva immunförsvaret som har förmåga att tidigt identifiera och oskadliggöra virusinfekterade celler.
Vid svår covid-19 har även färre cirkulerande dendritiska celler än hos friska kontroller [25], liksom samband mellan förekomst av omogna monocyter och sjukdomsförlopp [26], rapporterats. Såväl basofila som eosinofila granulocyter uppvisar en aktiverad fenotyp och har rapporterats vara involverade i utvecklingen av koagulationsrubbning och vävnadsskada vid svår covid-19 [27].
Koagulation
Rubbningar i koagulationssystemet är vanliga vid covid-19, och incidensen av tromboemboliska komplikationer ökar med sjukdomens svårighetsgrad [28]. I en fransk studie av 135 akutpatienter med covid-19 hade 15 procent DT-verifierad lungembolisering redan vid ankomst till sjukhus [29]. Lungembolisering eller djup ventrombos har rapporterats föreligga hos ungefär en fjärdedel av intensivvårdade patienter med covid-19 trots profylaktisk antikoagulation [28]. Många patienter med covid-19 uppvisar tidigt i förloppet tecken på koagulationsaktivering med höga nivåer av D-dimer och trombocytopeni [30]. Samband har vid covid-19 påvisats mellan patologiska koagulationsmarkörer, inklusive hyperreaktiva trombocyter, vid inskrivning på sjukhus och sjukdomens svårighetsgrad och mortalitet [31, 32].
Möjligen bidrar sars-cov-2 även till covid-19-associerad koagulopati via direkt trombocytaktivering, mot bakgrund av att trombocyter uttrycker såväl ACE-2 (angiotensin converting enzyme 2) som membranproteinet transmembranserinproteas-2, där viruset in vitro rapporterats binda in till och aktivera trombocyter [33]. Patienter med svår covid-19 har även rapporterats ha signifikant högre nivåer av, samt cirkulerande mikrovesikler med, TF (tissue factor, koagulationsfaktor III) än friska kontroller. Dessa, inklusive TF-uttryckande trombocyter, kan fungera som cirkulerande mediatorer av koagulopati [34]. Vidare föreligger direkta orsakssamband mellan inflammation, aktiverade monocyter och koagulationsaktivering inom ramen för konceptet immuntrombos. Cirkulerande proinflammatoriska molekyler (viruspartiklar, endogena vävnadspartiklar samt cytokiner) aktiverar monocyter till uttryck av TF och frisättning av mikrovesikler med membranbunden TF, vilket kan bidra till att utlösa en koagulationskaskad [35].
Endotel
En förutsättning för homeostas i koagulationssystemet är ett intakt och låginflammatoriskt endotel. Även endotelceller, som i hög grad uttrycker ACE-2, kan direkt infekteras med sars-cov-2. Aktivering med destruktion av endotel kan utlösa en koagulationskaskad genom uttryck av TF (monocyter) och rekrytering av immunceller (neutrofila granulocyter) [35, 36]. Graden av endotelskada in vivo kan uppskattas utifrån antalet cirkulerande endotelceller: högre nivåer vid covid-19 korrelerar med förekomst av tromboembolism och histopatologiskt påvisad endotelskada, och lägre nivåer med klinisk förbättring [37, 38]. Frisättning av proinflammatoriska cytokiner från vävnadbundna makrofager och mastceller, utlöst av molekylära signalmönster i närvaro av patogener vid inträffad vävnadsskada, kan få endotelceller att byta från en antiinflammatorisk och antikoagulatorisk fenotyp till en proinflammatorisk och prokoagulatorisk fenotyp med högre uttryck av adhesionsmolekyler och högre membranpermeabilitet [10, 39]. I lungbiopsier från patienter med covid-19-associerad ARDS har histopatologiskt markant mer utbredda endotelskador påvisats än vid ARDS i samband med H1N1-pneumonit eller bakteriell pneumoni [22, 40]. Utbredda endotelförändringar har föreslagits kunna förklara den långa raden av extrapulmonella kliniska fynd vid svår covid-19 och karakteriserats som specifika för covid-19 [23].
Komplement
En grundläggande del av det medfödda immunförsvaret utgörs av komplementsystemet. Genom att opsonisera virus och uppreglera interferonsignalering bidrar komplementsystemet till att kontrollera en virusinfektion, men samtidigt kan det skada kroppens egna vävnader om det inte kontrolleras. Vid hyperinflammatoriska tillstånd förändras interaktionen mellan komplement, immunceller och koagulation. Neutrofila granulocyter svarar sämre på anafylatoxiner, makrofager frisätter mer cytokiner och endotelceller uttrycker TF på cellytan, vilket ökar risken för systemiska inflammations- och koagulationskaskader och därmed även för omfattande vävnadsskada [41].
Hos sars-cov-2 interagerar ett strukturellt nukleokapsidprotein (N-protein) med det nyckelenzym som aktiverar komplementsystemet, mannanbindande lektinassocierat serinproteas-2, och förstärker dess effekter [42]. Komplementfaktor C5b, en slutprodukt i komplementkaskaden som förekommer bronkoalveolärt vid svår covid-19, verkar som ett anafylatoxin med mediering av kemotaxi, frisättning av cytokiner, ROS och NET samt degranulering av immunceller. Vid både sars och mers anses C5b ha haft betydelse för utveckling av ARDS [43]. I histopatologiskt material av lungvävnad från patienter som avled i covid-19 sågs aktivering av såväl komplement som signalvägar både i alveoler och i lungans blodkärl [44]. I en retrospektiv amerikansk kohortstudie rapporterades ingen covid-19-patient med komplementbristsjukdom (en oberoende riskfaktor för svår infektionssjukdom) ha utvecklat allvarlig sjukdom, i motsats till patienter med åldersrelaterad makuladegeneration (en surrogatmarkör för komplementaktivering), som oftare utvecklade svår covid-19 [45].
Lungfunktion
Mikrotrombotisering i lungorna är vanlig vid covid-19-associerad ARDS, där endotelskador sannolikt är patofysiologiskt starkt bidragande [4, 46]. Av 334 obducerade covid-19-patienter hade över 90 procent tecken på mikrotrombotisering, huvudsakligen i lungorna, med trombocyt- och fibrinnätverk associerade med NET, i enlighet med konceptet immuntrombos [4]. Trombotisering i lungornas kapillärsystem vid svår covid-19 med ARDS försämrar balansen mellan ventilation och lungperfusion, vilket leder till ökad pulmonell shuntning, ökad funktionell »dead space«-ventilation och syresättningsproblem. Trombolytisk infusion av vävnadsplasminogenaktivator (tPA) har rapporterats ge övergående förbättring av syresättningen vid svår covid-19 med ARDS [5], men risken för blödningsbiverkningar är betydande vid systemisk tillförsel.
I lungvävnad från patienter avlidna med covid-19-associerad ARDS korrelerar inte mängden viruspartiklar med omfattningen av inflammatorisk skada, då diffus alveolär skada (DAD) och bronkopneumoni föreligger såväl i områden med som utan detekterbar sars-cov-2-arvsmassa [47]. Detta talar starkt för att även andra mekanismer än direkt virusmedierade driver patogenesen vid svår covid-19. Denna dissociation mellan inflammation och virusnärvaro i lungorna, som kan spegla kvardröjande inflammatorisk aktivitet efter att viruset eliminerats och/eller inflammation i områden utan virusreplikation, är tecken på ett onormalt fungerande och överdrivet inflammatoriskt svar. Liksom vid ARDS av annan etiologi föreligger ödem i och skador på alveolerna vid svår covid-19 [4]. Även lågt serumalbumin vid svår covid-19 har rapporterats korrelera med röntgenologiskt mer utbredd lungskada och sämre 30-dagarsöverlevnad [40].
Diskussion
Covid-19-pandemin är fortfarande långt ifrån över, även om den pågående massvaccinationen förhoppningsvis innebär början på slutet för en sedan andra världskriget aldrig skådad världsomfattande humanitär kris. Globalt suboptimal fördelning av tillgängligt vaccin, tillkomst av nya virusvarianter, sällsynta men allvarliga vaccinbiverkningar samt lavinartade smittvågor i länder med låg vaccintäckning och otillräckliga smittskyddsåtgärder kan komma att fortsatt utmana människor, sjukvårdssystem och politiker i alla världsdelar under lång tid framöver.
Lungorna har såväl anatomiska som fysiologiska förutsättningar att ta stor skada vid covid-19, samtidigt som de har en fundamental roll för individens överlevnad. Aktivering av det kraftfulla medfödda immunförsvaret i lungorna anses kunna bidra till vävnadsskada och utveckling av ARDS och immuntrombos. Inflammatoriskt medierat alveolärt ödem leder, tillsammans med virusutlösta och immunologiskt orsakade kapillära endotelskador i lungorna med mikroangiopatier och mikrotrombotisering, till ventilations-/perfusionsobalans med pulmonell shuntning och arteriell hypoxemi. Samtidig förekomst av lungembolisering, som ytterligare försämrar lungornas förmåga till syresättning, bör differentialdiagnostiskt övervägas vid svår hypoxemi hos patienter med covid-19.
Det oproportionerliga immunsvaret vid svår covid-19 verkar vara drivande i patogenesen. Den brittiska RECOVERY-studien har som första studie övertygande visat att immunmodulerande behandling med glukokortikosteroider vid behov av andningsunderstödjande behandling inklusive syrgas förbättrar överlevnaden med covid-19 på sjukhus [48]. Samma forskargrupp har även rapporterat, om än mindre övertygande, att tillägg av IL-6-hämmare ytterligare sänker mortaliteten vid svår covid-19 [49]. Även motsvarande stöd för behandling med IL-1-hämmare har rapporterats [50].
En större fransk kohortstudie av sjukhusvårdade covid-19-patienter talar för två övergripande sjukdomsfaser – en initial med högre virustitrar och lägre inflammatorisk aktivitet följd av en senare med lägre virustitrar och högre inflammatorisk aktivitet. Baserat på datamodellering skulle maximala virustitrar ha förelegat redan något dygn före symtomdebuten, och ju längre tid därefter som virus fortsatt kunde påvisas, desto högre var mortaliteten [51]. Mycket talar för att immunförsvaret vid svår covid-19 inte klarar av att eliminera viruset, vilket i sin tur leder till ett dysfunktionellt och skadligt svar på infektionen [23]. Under sjukdomsförloppet minskar alltså graden av direkt virusmedierad vävnadsskada samtidigt som graden av indirekt inflammatoriskt medierad vävnadsskada ökar.
Antiviral behandling ska i förekommande fall ges tidigt i det kliniska förloppet för att begränsa virusmedierad cytotoxicitet och öka immunförsvarets möjligheter att bekämpa infektionen, men behandlingseffekter är svåra att i efterhand tillskriva sådan behandling, då det fortsatta sjukdomsförloppet initialt är omöjligt att förutse i det enskilda fallet. Inväntar man å andra sidan svår covid-19 innan man påbörjar antiviral behandling kan värdet ifrågasättas.
Immunmodulerande behandling är ännu svårare att använda. Vid tillförsel tidigt eller sent i sjukdomsförloppet uppväger inte den möjliga nyttan riskerna. Att tidigt i förloppet hämma immunförsvaret skulle teoretiskt kunna förlänga tiden till virusfrihet och därmed öka risken för virusmedierad vävnadsskada. Att i stället avvakta med sådan behandling tills ett kraftigt immunologiskt svar utvecklats ökar risken för immunologiskt medierad vävnadsskada. Vid en svårbestämbar, och för varje patient unik, tidpunkt med minimal pågående (avtagande) virusmedierad och (tilltagande) inflammatoriskt medierad cytotoxisk vävnadspåverkan under covid-19 skulle man alltså teoretiskt kunna förvänta sig störst nytta av, och minst risker med, immunmodulerande behandling.
Slutsatser
Patofysiologin vid svår covid-19 inbegriper ett flertal samverkande mekanismer utöver direkt virusmedierad vävnadsskada. Ett bristfälligt reglerat initialt immunförsvar, medierat via såväl cellulära som molekylära mekanismer, utlöser en självförstärkande spiral av okontrollerad parallell inflammations- och koagulationsaktivering, vilket med samtidig aktivering av komplementsystemet kan orsaka utbredda skador på alveoler och endotel i lungorna. Skador på i första hand lungkapillärernas endotel, i andra hand kapillärendotel i systemkretsloppet, stimulerar koagulationssystemet ytterligare, vilket dels bidrar till mer massiv mikrotrombotisering med ännu sämre syresättning, och dels förstärker det systeminflammatoriska svaret. Störst nytta av immunmodulerande behandling kan förväntas vid samtidiga tecken på avtagande virusinfektion och tilltagande inflammation.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
(uppdaterad 2022-12-01)
Referenser
- Grant MC, Geoghegan L, Arbyn M, et al. The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19): a systematic review and meta-analysis of 148 studies from 9 countries. PLoS One. 2020;15(6):e0234765.
- Du Y, Tu L, Zhu P, et al. Clinical features of 85 fatal cases of COVID-19 from Wuhan. A retrospective observational study. Am J Respir Crit Care Med. 2020;201(11):1372-9.
- Jiménez D, García-Sanchez A, Rali P, et al. Incidence of VTE and bleeding among hospitalized patients with coronavirus disease 2019: a systematic review and meta-analysis. Chest. 2021;159(3):1182-96.
- Chen W, Pan JY. Anatomical and pathological observation and analysis of SARS and COVID-19: microthrombosis is the main cause of death. Biol Proced Online. 2021;23(1):4.
- Poor HD, Ventetuolo CE, Tolbert T, et al. COVID-19 critical illness pathophysiology driven by diffuse pulmonary thrombi and pulmonary endothelial dysfunction responsive to thrombolysis. Clin Transl Med. 2020;10(2):e44.
- Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718-24.
- Del Valle DM, Kim-Schulze S, Hsin-Hui H, et al. An inflammatory cytokine signature helps predict COVID-19 severity and death. Medrxiv. Epub 30 maj 2020. doi: 10.1101/2020.05.28.20115758.
- Sinha P, Matthay MA, Calfee CS. Is a »cytokine storm« relevant to COVID-19? JAMA Intern Med. 2020;180(9):1152-4.
- Chiang CC, Korinek M, Cheng WJ, et al. Targeting neutrophils to treat acute respiratory distress syndrome in coronavirus disease. Front Pharmacol. 2020;11:572009.
- Preissner KT, Fischer S, Deindl E. Extracellular RNA as a versatile DAMP and alarm signal that influences leukocyte recruitment in inflammation and infection. Front Cell Dev Biol. 2020;8:619221.
- Zimmermann M, Arruda-Silva F, Bianchetto-Aguilera F, et al. IFNα enhances the production of IL-6 by human neutrophils activated via TLR8. Sci Rep. 2016;6:19674.
- Guihot A, Litvinova E, Autran B, et al. Cell-mediated immune responses to COVID-19 infection. Front Immunol. 2020;11:1662.
- Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11):e138999.
- Zemans RL, Matthay MA. What drives neutrophils to the alveoli in ARDS? Thorax. 2017;72(1):1-3.
- Veras FP, Pontelli MC, Silva CM, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. 2020;217(12):e20201129.
- Thierry AR, Roch B. Neutrophil extracellular traps and by-products play a key role in COVID-19: pathogenesis, risk factors, and therapy. J Clin Med. 2020;9(9):2942.
- Page EM, Ariëns RAS. Mechanisms of thrombosis and cardiovascular complications in COVID-19. Thromb Res. 2021;200:1-8.
- Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169-79.
- Morena V, Milazzo L, Oreni L, et al. Off-label use of tocilizumab for the treatment of SARS-CoV-2 pneumonia in Milan, Italy. Eur J Intern Med. 2020;76:36-42.
- Yang Q, Xie L, Zhang W, et al. Analysis of the clinical characteristics, drug treatments and prognoses of 136 patients with coronavirus disease 2019. J Clin Pharm Ther. 2020;45(4):609-16.
- Mahale N, Rajhans P, Godavarthy P, et al. A retrospective observational study of hypoxic COVID-19 patients treated with immunomodulatory drugs in a tertiary care hospital. Indian J Crit Care Med. 2020;24(11):1020-7.
- Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med. 2020;383(2):120-8.
- Osuchowski MF, Winkler MS, Skirecki T, et al. The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med. 2021;9(6):622-42.
- Maucourant C, Filipovic I, Ponzetta A, et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci Immunol. 2020;5(50):eabd6832.
- Kvedaraite E, Hertwig L, Sinha I, et al; Karolinska KI/K COVID-19 Study Group. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc Natl Acad Sci U S A. 2021;118(6):e2018587118.
- Silvin A, Chapuis N, Dunsmore G, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2020;182(6):1401-18.e18.
- Lourda M, Dzidic M, Hertwig L, et al; Karolinska KI/K COVID-19 Study Group. High-dimensional profiling reveals phenotypic heterogeneity and disease-specific alterations of granulocytes in COVID-19. Proc Natl Acad Sci USA. 2021;118
(40):e2109123118. - Frazer JS, Tyrynis Everden AJ. Emerging patterns of hypercoagulability associated with critical COVID-19: a review. Trends in Anaesthesia and Critical Care. 2020;34:4-13.
- Jevnikar M, Sanchez O, Andronikov M, et al. Prevalence of pulmonary embolism in patients at the time of hospital admission for COVID-19 [abstrakt PB/CO10]. International Society on Thrombosis and Haemopstasis (ISTH) congress, 12–14 jul 2020.
- Rahi MS, Jindal V, Reyes SP, et al. Hematologic disorders associated with COVID-19: a review. Ann Hematol. 2021;100(2):309-20.
- Tang N, Li D, Wang X, et al. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844-7.
- Comer SP, Cullivan S, Szklanna PB, et al; COCOON Study investigators. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021;19(2):e3001109.
- Sahai A, Bhandari R, Koupenova M, et al. SARS-CoV-2 receptors are expressed on human platelets and the effect of aspirin on clinical outcomes in COVID-19 patients. Res Sq. 2020;rs.3.rs-119031.
- Canzano P, Brambilla M, Porro B, et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl Sci. 2021;6(3):202-18.
- Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Revs Immunol. 2020;20(6):355-62.
- McFadyen JD, Stevens H, Peter K. The emerging threat of (micro)thrombosis in COVID-19 and its therapeutic implications. Circ Res. 2020;127(4):571-87.
- Diehl JL, Peron N, Chocron R, et al. Respiratory mechanics and gas exchanges in the early course of COVID-19 ARDS: a hypothesis-generating study. Ann Intensive Care. 2020;10(1):95.
- Rambaldi A, Gritti G, Micò MC, et al. Endothelial injury and thrombotic microangiopathy in COVID-19: Treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology. 2020;225(6):152001.
- Gozzo L, Viale P, Longo L, et al. The potential role of heparin in patients with COVID-19: beyond the anticoagulant effect. A review. Front Pharmacol. 2020;11:1307.
- Wu MA, Fossali T, Pandolfi L, et al. Hypoalbuminemia in COVID-19: assessing the hypothesis for underlying pulmonary capillary leakage. J Intern Med. 2021;289(6):861-72.
- Lo MW, Kemper C, Woodruff TM. COVID-19: complement, coagulation, and collateral damage. J Immunol. 2020;205(6):1488-95.
- Flude BM, Nannetti G, Mitchell P, et al. Targeting the complement serine protease MASP-2 as a therapeutic strategy for coronavirus infections. Viruses. 2021;13(2):312.
- Ram Kumar Pandian S, Arunachalam S, Deepak V, et al. Targeting complement cascade: an alternative strategy for COVID-19. 3 Biotech. 2020;10(11):479.
- Rendeiro AF, Ravichandra H, Bram Y, et al. The spatial landscape of lung pathology during COVID-19 progression. Nature. 2021;593(7860):564-9.
- Ramlall V, Thangaraj PM, Meydan C, et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat Med. 2020;26(10):1609-15.
- Nuche J, de la Cal TS, Guarch CJL, et al. Effect of coronavirus disease 2019 in pulmonary circulation. The particular scenario of precapillary pulmonary hypertension. Diagnostics (Basel). 2020;10(8):548.
- Dorward DA, Russell CD, Um IH, et al. Tissue-specific immunopathology in fatal COVID-19. Am J Respir Crit Care Med. 2021;203(2):192-201.
- RECOVERY Collaborative Group; Horby P, Lim WS, Emberson JR, et al. Dexamethasone in hospitalized patients with covid-19. N Engl J Med. 2021;384(8):693-704.
- RECOVERY Collaborative Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397(10285):1637-45.
- Kyriakoulis KG, Kollias A, Poulakou G, et al. The effect of anakinra in hospitalized patients with COVID-19: an updated systematic review and meta-analysis. J Clin Med. 2021;10(19):4462.
- Néant N, Lingas G, Le Hingrat Q, et al; French COVID Cohort Investigators and French Cohort Study groups. Modeling SARS-CoV-2 viral kinetics and association with mortality in hospitalized patients from the French COVID cohort. Proc Natl Acad Sci U S A. 2021;118(8):e2017962118.
Summary
Severe Covid-19 has a high mortality rate. Vital organ dysfunction results from pathophysiological self-amplifying loops of innate immunological hyperactivation, cytokine release, complement deposition, endothelial damage, and macro- and microvascular thromboembolism. Resulting alveolar exudation and pulmonary capillary thromboembolism lead to ventilation-perfusion mismatch, considered to be a primary cause of death in severe Covid-19. Therapeutic immunomodulation is believed to be safer and more effective during time periods with decreasing viral exposition and increasing inflammation.
Vänliga hälsningar
Jonas Åkeson
Den mekanism du beskriver skulle delvis kunna förklara den kraftiga försämring med allvarlig lungpåverkan som noterats hos vissa patienter med svår covid-19 och kliniska tecken på pneumoni/sepsis, men vi bedömer att ömsesidig positiv interaktion mellan ökad inflammation och ökad koagulation kvarstår som en grundläggande förklaringsmodell till klinisk försämringen hos våra svårast sjuka koronapatienter.