IgA-nefropati (IgAN) utgör majoriteten av alla glomerulonefriter.
30–40 procent av patienter med IgAN utvecklar terminal njursvikt.
Minst en fjärdedel av alla patienter som behöver dialys har glomerulonefrit som underliggande sjukdom.
Symtom och klinik är initialt diskreta, varför det är viktigt att känna till sjukdomen.
Den kliniska bilden vid debuten utgörs av mikro- eller makroskopisk hematuri, ibland albuminuri, varierande grad av progressiv njurskada och vanligen hypertoni.
I dag saknas tillförlitliga prognostiska biomarkörer, men forskning pågår.
Tidig diagnos, adekvat intervention och uppföljning är viktigt för att fördröja, och om möjligt förhindra, behandlingskrävande njursvikt.
Var fjärde patient som utvecklar terminal njursvikt med dialys- och transplantationsbehov har glomerulonefrit som uremiorsakande sjukdom (Fakta 1). IgA-nefropati (IgAN) är den vanligaste formen av glomerulonefrit i världen, och utvecklas i cirka 30–40 procent av fallen till en terminal uremi. Den globala incidensen är cirka 2,5/100 000/år för vuxna. Prevalensen varierar geografiskt. Den kaukasiska befolkningen har en 5 gånger högre risk och den asiatisk-amerikanska en 15-faldigt ökad risk att utveckla en behandlingskrävande njursvikt på grund av IgAN jämfört med afro-amerikaner [1]. Utöver detta har, enligt Svenskt njurregister, 10 procent av alla dialyspatienter hypertoni och 23 procent »övriga sjukdomar« som uremiorsakande åkomma. Det finns anledning att misstänka att det döljer sig fler patienter med IgAN i dessa två kategorier, eftersom diagnostisk njurbiopsi saknas [2].
Debutsymtom vid IgAN är varierande, men inte sällan har patienten en mikroskopisk hematuri vid en infektion i till exempel luftvägarna eller mag–tarmkanalen. 10–15 procent utvecklar makroskopisk hematuri, vilket medför att patienten söker sjukvård. Tyvärr resulterar makroskopisk hematuri ofta i att enbart en urologisk (och inte njurmedicinsk) utredning bedrivs, där normala undersökningsresultat innebär att patienten »friskförklaras«. Detta är särskilt beklagligt när det gäller unga personer där vi vet att en tidig diagnos och interventioner kan bromsa förloppet av IgAN. Hypertoni förekommer hos 30 procent och albuminuri hos 46 procent i tidigt skede av IgAN [3].
Det finns en systemisk variant av IgAN, IgA-vaskulit (tidigare kallad Henoch–Schönlein-purpura) som utöver njurengagemang ger inflammation i små blodkärl i hud och mag–tarmkanal (framför allt tunntarm). Kärlinflammationen leder till palpabel purpura, framför allt på nedre extremiteter där hudbiopsi visar en leukocytoklastisk vaskulit. Med immunfluorescens kan ofta IgA-inlagring påvisas i kärlväggen. I vissa fall förekommer även sår i gastrointestinalkanalen med buksmärta och eventuell blodig diarré samt artralgi och myalgi.
Förekomst av palpabel purpura [4] är enligt nuvarande klassifikation huvudkriterium för IgA-vaskulit (IgAV). I en studie på 260 vuxna patienter med IgAV hade 70 procent glomerulonefrit och 53 procent gastrointestinala symtom. Kronisk njursvikt utvecklas hos 11–30 procent [5]. IgAV är vanligare hos barn än vuxna och har då också oftast en gynnsam prognos. IgAV och IgAN kan vara associerade med andra sjukdomar som IBD, ankyloserande spondylit och leversjukdomar. Både isolerad hudvaskulit och IgAN kan utvecklas till systemisk IgA-vaskulit [6].
Diagnostik
- Diagnos av IgAN kan endast ställas med njurbiopsi.
- Njurbiopsi ska graderas enligt Oxford MEST-C classification.
- Det finns i dag inga serologiska diagnostiska test för IgAN.
- Alla patienter med IgAN måste utredas avseende eventuell bakomliggande orsak till nefropatin, det vill säga att man identifierar sekundära former.
- I urinsediment kan korniga cylindrar påträffas samtidigt med mikroskopisk hematuri, framför allt om urinen undersöks i anslutning till inflammation eller infektion. Vid IgAN-skov i samband med infektion finns ibland symtom av urinträngningar (uretritsymtom men avsaknad av bakteriuri) eller diffus tyngdkänsla med värk i ryggen över njurlogerna. Albuminuri eller sänkt GFR (glomerulär filtrationshastighet) stärker indikationen för njurbiopsi. Njurbiopsi är standard för påvisande av IgA-depositioner tillsammans med komplementfaktor C3 i glomerulis mesangieområden.
Man ska definiera och klassificera IgAN med hjälp av den så kallade Oxfordklassifikationen. Denna baseras på fyra histopatologiska kännetecken vid IgAN, så kallade MEST-poäng: mesangial (M), endokapillär hypercellularitet (E), segmental glomeruloskleros (S) och tubulär atrofi/interstitiell fibros (T). Sedermera har ett femte fynd, crescents (C) (halvmånar) inkluderats i bedömningen [7, 8].
Patogenes och risk för utveckling av IgAN
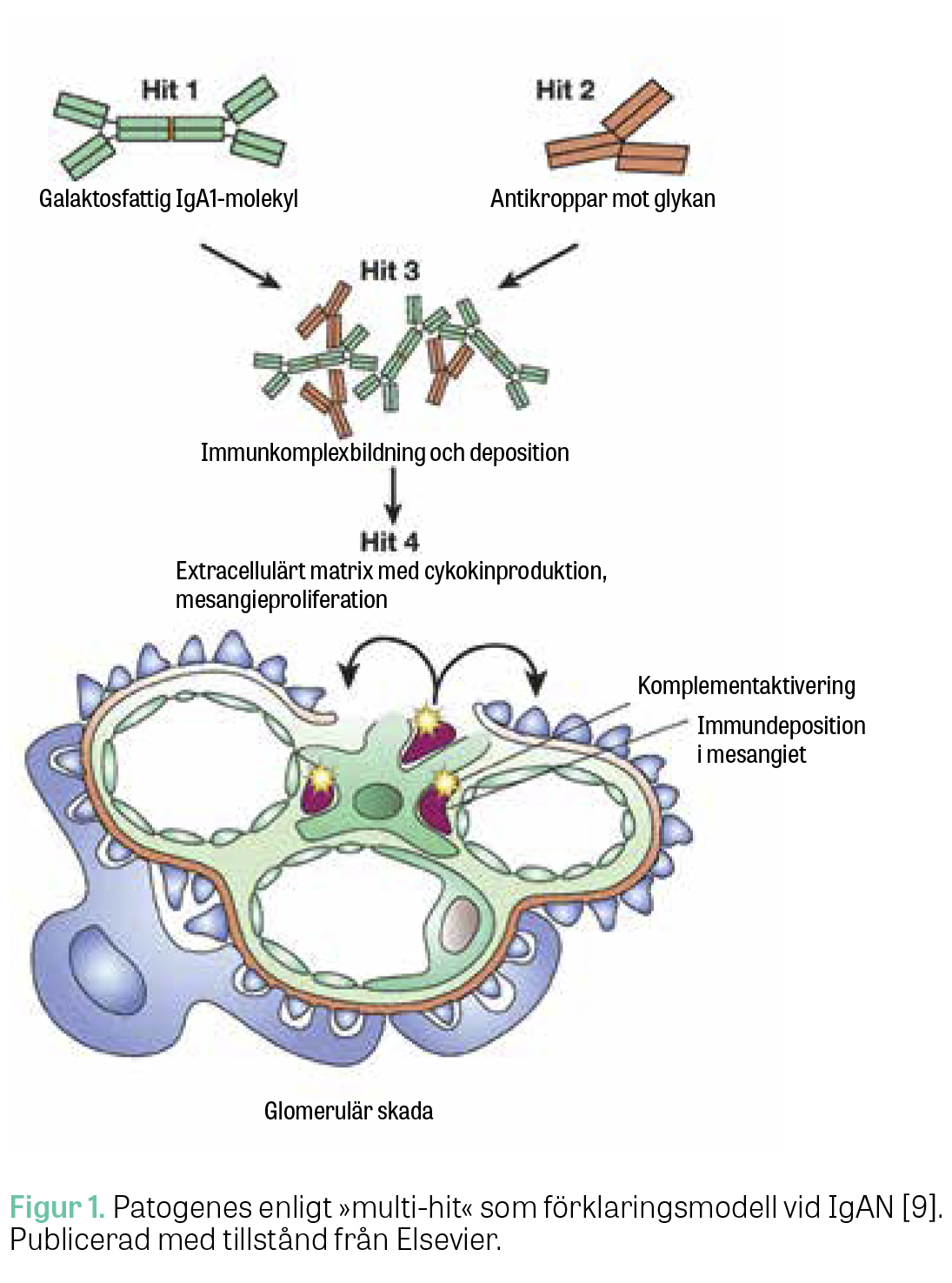
Risken att utveckla IgAN påverkas av olika genetiska faktorer och miljöfaktorer. Patogenesen beskrivs enligt nuvarande kunskap som en flerstegsprocess (»multi-hit-process«), nedan beskriven i 4 steg [9] (Figur 1).
Steg 1: Avvikande produktion av IgA. Patienter med IgAN har visat sig bilda avvikande IgA1-antikroppar (galaktosfattigt IgA1, Gd-IgA1) som tenderar till att forma större immunkomplex, vilka undgår nedbrytning i levern. Bildning av IgA1 sker i B-lymfocyter i peyerska plack, tonsiller och benmärg och resulterar i stora immunkomplex med antiglykan-IgG som hamnar i blodbanan [10]. Galaktosfattigt IgA1 (Gd-IgA1) uppstår genom en defekt i glukosylering vid produktionen av IgA1-antikroppar. Glukosyleringsdefekten uppstår oftast i polymeriskt IgA1 som produceras av IgA1-utsöndrande lymfocyter som sitter i anslutning till mukosan i till exempel peyerska plack och tonsiller. Det är oklart hur IgA1 kommer ut till cirkulationen hos patienter med IgAN. En teori är att IgA1-producerande B-lymfocyter vandrar in till benmärgen på grund av förändrat uttryck av målsökande receptorer (»homing-receptorer«) på cellytan. En annan teori är att en antigenstimulering leder till en ökad reaktivitet i mukosans lymfocyter som i sin tur producerar för mycket IgAN, som sedan läcker ut till cirkulationen [9].
Steg 2: Antikroppar mot Gd-IgA1. Höjda nivåer av Gd-IgA1 kan leda till produktion av autoantikroppar mot just Gd-IgA1. Det finns två olika autoantikroppar: IgG- respektive IgA-klass. Antikropparna verkar vara förknippade med en ökad risk för sjukdomsprogress, dialysbehov och död [11]. IgG-anti-Gd-IgA1 kunde påvisas hos patienter med IgAN med 89 procents sensitivitet och 92 procents specificitet [12]. Kliniska långtidsstudier saknas.
Steg 3: Ansamling av immunkomplex som fastnar i glomeruli. Ovan beskrivna autoantikroppar bildar tillsammans med Gd-IgA1 cirkulerande immunkomplex som fastnar i glomeruli, framför allt i mesangiet, och stimulerar där en lokal cellproliferation och frisättning av inflammatoriska mediatorer som resulterar i proteinuri, fibros och på sikt njursvikt.
Steg 4: Inflammation och komplementaktivering. Immunkomplexen i glomerulärt mesangium aktiverar komplementsystemet, stimulerar mesangiecellerna och inducerar sekretion av cytokiner, kemokiner och extracellulära matrixproteiner. Tillsammans verkar de proinflammatoriskt och profibrotiskt i glomeruli [9]. Kvoten mellan Gd-IgA1 och plasmanivån av komplementfaktor C3 är associerad med progress av kronisk njursvikt vid IgAN [13]. Dysreglering av komplementsystemet, framför allt med komplementfaktor H-relaterade proteiner 1 och 5 och av den lektinrelaterade vägen, verkar vara förknippade med allvarligare sjukdomsförlopp av IgAN [14-16]. Låga nivåer av membranbundna komplementinhibitorer som CD46 och CD55 har visats vara associerade med en sämre prognos vid IgAN, i tillägg till MEST-C-poäng och kliniska variabler som kombinationspaket för att vässa prognos med MEST-C [17]. Höga nivåer av CD46 däremot hämmar komplementaktivitet och skulle därmed kunna bromsa sjukdomsprogressen [17].
Ärftlighet och omgivningsfaktorer
Ärftlighet kan predisponera för utveckling av IgAN, och studier har påvisat multipla riskalleler [18]. Upp till 15 procent av alla IgAN-fall har en familjär koppling, definierat som att åtminstone 2 personer med diagnostiserad IgAN finns inom en och samma släkt [19].
Genomtäckande associationsstudier (genome-wide association study [GWAS]) för IgAN har genomförts med 5 stora fall–kontrollstudier [20]. Kiryluk et al fann att vissa GWAS-lokus för IgAN kodar för många olika proteiner som spelar roll för det medfödda immunförsvaret, bland annat komplementaktivering, bevarande av intestinal slemhinnebarriär och slemhinnors IgA-produktion. Gemensamma genetiska lokus har identifierats för IgAN och framför allt andra autoimmuna sjukdomar som till exempel SLE och inflammatoriska tarmsjukdomar [21].
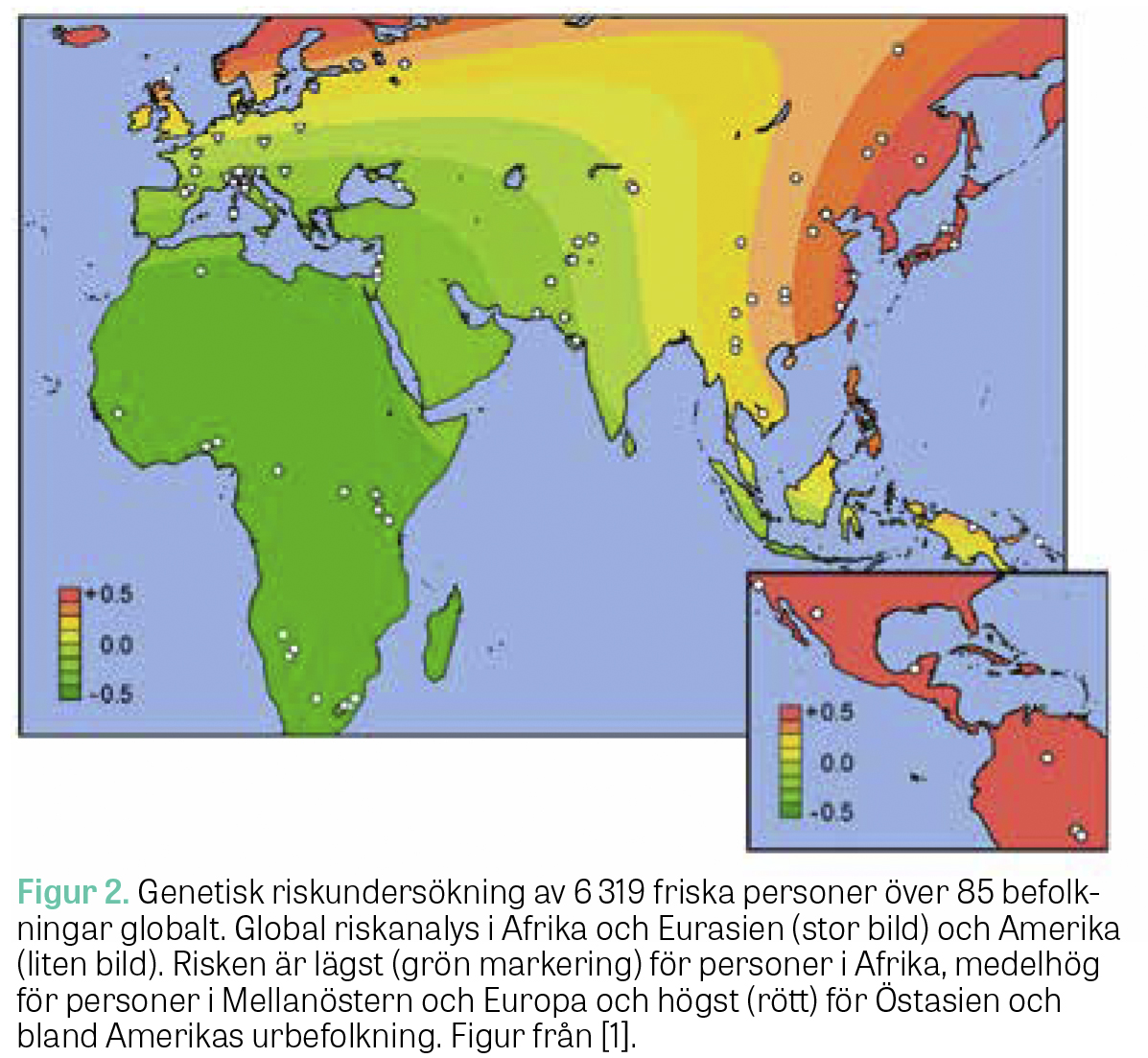
Genomtäckande associationsstudier kunde även visa att den genetiska risken för IgAN varierar med samma mönster som den geografiska skillnaden i sjukdomsprevalensen [1]. Vid tilltagande avstånd från Afrika ökar den genetiska predispositionen för IgAN med en signifikant väst–öst- och syd–nord-riskgradient.
Geografiskt minskad risk för IgAN har spekulerats bero på ett genetiskt skydd mot regionalt förekommande maskinfektioner, som trots förbättrad folkhälsa är ett vanligt förekommande problem i Afrika och Östasien [20–23] (Figur 2).
Behandling
I första hand rekommenderas behandling med syfte att minska proteinurin, vilket reducerar risken för att utveckla njursvikt. Intensiv behandling av proteinurin framför allt med RAAS (renin–angiotensin–aldosteronsystemet)-blockerare rekommenderas [24], men även behandling med SGLT-2-hämmare har i studier publicerade under det senaste året visat sig minska proteinurin [25].
Viktreduktion och rökstopp kan ge ytterligare resultat vid behandling av proteinuri. Även behandling med statiner har beskrivits resultera i en minskning av proteinurin enligt en liten studie med 21 patienter [26].
Kortikosteroidbehandling i 6 månader har i flera studier påvisat en reduktion av proteinuri och en förbättring av njuröverlevnad [27]. I Pozzis randomiserade studie från 1999 [28] deltog patienter med biopsiverifierad IgAN och signifikant proteinuri samt kreatinin <133 µmol/l, där behandlingsgruppen fick prednisolon under 6 månader. Långtidsuppföljning visade en god effekt med 97 procent njuröverlevnad för kortisonbehandlade jämfört med 53 procent för kontrollgruppen [29]. Kritiken mot Pozzis studie var att endast 14 procent av patienterna i vardera gruppen hade fått RAAS-blockad.
Systemisk behandling med kortikosteroider kan dock bidra till allvarliga infektioner samt viktuppgång och diabetes. Enteralt verkande kortison (budesonid) med effekt i distala ileum, där det finns en hög förekomst av peyerska plack, har därför fått uppmärksamhet. NEFIGAN-studien genomfördes 2017 i 10 olika europeiska länder på 149 patienter med IgAN och kunde visa att 16 mg TRF (targeted-release formulation) av budesonid/dag som tillägg till RAAS-blockerare kunde minska albuminurin signifikant, utan att ge påtagliga systemiska biverkningar av kortisonbehandlingen [30]. Effekten, en nära 25-procentig minskning av proteinurin, kvarstod åtminstone 3 månader efter avslutad budesonidterapi.
Kombinationsterapi med kortikosteroider och cyklofosfamid eller azatioprin rekommenderas endast till patienter med aggressiv och progressiv sjukdom (snabbt förlöpande njursvikt, njurbiopsi med »crescents«). Studierna tyder på en viss förbättring i njuröverlevnad, men många biverkningar noterades [24]. Immunsuppressiva läkemedel i form av mykofenolsyra och rituximab har använts i aktiva svåra fall [31–33].
Det senaste tillskottet i terapi är komplementhämning, och flera randomiserade studier pågår [34].
Njurtransplantation är möjlig; recidiv av IgAN i transplantatet utvecklas dock hos en tredjedel av patienterna [35].
Prognos
30–40 procent av de patienter som har diagnostiserats med IgA-nefropati utvecklar en behandlingskrävande njursvikt inom loppet av 20 år.
Kända kliniska markörer som finns vid diagnostidpunkten och som medför en dålig prognos är hypertoni, proteinuri och nedsatt eGFR [36]. Proteinuri kan dock inte skilja mellan akut och kroniskt förlopp av IgAN [37].
I en studie från Italien visades att patienter med familjär form av IgAN har en sämre njuröverlevnad än de som har en sporadisk form; 20 års njuröverlevnad beräknas till 41 procent vid familjär jämfört med 94 procent vid sporadisk form; P = 0,003 [38].
Graviditet i sig verkar inte vara en riskfaktor för progress, förutsatt opåverkad GFR och frånvaro av eller låggradig albuminuri. En nyligen publicerad svensk registerstudie visade att gravida med IgAN har en ökad risk för preeklampsi samt förtidig födsel, varför särskild övervakning i specialistmödravården behövs [39]. Vid redan utvecklad måttlig njursvikt (minst CKD grad 3) beskrev Su et al 2017 snabbare progress av njursvikt [40].
IgA finns rikligt i tarmslemhinnans immunsystem, och man kan spekulera om huruvida försämring i det mikrobiotiska tarmsystemet kan vara en del av mekanismen bakom utveckling av IgAN [41].
En riskkalkylator, tillgänglig som applikation på mobilenhet, har nyligen presenterats av Barbour et al [42]. Denna kan vara till hjälp för att bedöma prognos, det vill säga risk att utveckla terminal njursvikt inom 5–7 år. Kalkylatorn inkluderar kliniska riskfaktorer som ålder, etnicitet, eGFR, blodtryck, grad av albuminuri, RAAS-blockad, MEST-poäng och eventuell erhållen immunhämmande behandling [42].
Biomarkörer vid IgAN
Idealiska biomarkörer bör kunna detektera subklinisk sjukdomsaktivitet och förutse sjukdomsförlopp, terapisvar samt recidivrisk av IgAN efter njurtransplantation [43]. Behovet av kontroller på njurmottagningen vid god prognos skulle i så fall minska. Likaså skulle behovet av immundämpande behandling kunna begränsas till de fall där man har indikation om en sämre prognos.
Vi gjorde en litteratursökning för perioden 1998–2018 med sökord »IgA nephropathy«, »biomarkers«, »autoantigens«, »autoantibodies«, »complement system proteins«, »risk factors«, »protein markers«, »low-molecular-weight proteins« och »prognostic factors«. Ett urval gjordes baserat på publicerade studier med minst 5 års uppföljningstid på primär IgAN hos vuxna individer.
Sammanfattningsvis föreföll den mest undersökta biomarkören vara Gd-IgA1 med en sensitivitet på 56–76 procent och specificitet på 89–94 procent för diagnos av IgAN [12, 44, 45]. Denna markör kan även detekteras hos friska anhöriga till patienter med IgAN [46]. IgG-antikroppar mot Gd-IgA1 kan också ha betydelse, men kanske mer för att studera patogenes. Fokus i dag ligger mycket på komplementsystemet i patogenesen av IgAN [47].
Litteraturgenomgången kunde inte identifiera några andra stabila biomarkörer utöver de tidigare kända, som utgörs av patientens kön, ålder, njurfunktion respektive proteinurigrad vid tidpunkten för IgAN-diagnos. Dock kan nämnas MMP-7 (matrixmetalloproteinas-7), ACE-polymorfi, Fc-receptor för komplement; genotyp FcγIIIb och CD14/-159-polymorfi, vilka kan spela en roll för utveckling och recidivrisk av IgAN.
Diskussion
Efter genomgång av litteraturen angående biomarkörer för IgAN kan vi konstatera att det pågår intensiv forskning kring ämnet, samtidigt som man ännu inte har lyckats identifiera någon solid och valid biomarkör, undantaget Gd-IgA1. Det finns fortfarande ingen bra biomarkör som kan ersätta njurbiopsi för att ställa diagnosen hos annars friska unga människor. Inte heller har man kunnat hitta en biomarkör som kan underlätta valet av behandlingsstrategi.
En aktuell litteraturgranskning bekräftade endast redan kända markörer för njursvikt som till exempel kreatinin, hematuri, albuminuri, hypoalbuminemi och en från början nedsatt njurfunktion. Risken att utveckla terminal njursvikt är självklart högre om patienten tidigt har en sviktande funktion. Ålder som riskfaktor i sig medför en reduktion av eGFR och därmed sannolikt en sämre prognos, vilken dock inte är specifik för IgAN. Empiriska studier av bland andra Wakai kring ohälsosamt matintag och progression av IgAN belyser ett möjligt samband mellan mikrobiotisk tarmhälsa och progress av njursjukdomen [48].
En biomarkör som kan vara lovande är MMP-7 (matrixmetalloproteinas-7) i blodprov avseende risk för njursvikt [49], men mer forskning med fler patienter behövs för att bekräfta dess funktion som prognostisk biomarkör. Tilläggas kan att MMP-7 är associerad med glomerulär skleros och tubulointerstitiell fibros, vilket också speglas av att den histologiska bilden i T1-2 i MEST-C har betydelse för prognosen.
Genetisk predisposition har betydelse för uppkomst av IgAN, och etnicitet spelar en roll vad gäller sjukdomsprognosen. I Sverige, där vi har många patienter från olika delar av världen, är det därför angeläget att beakta IgAN vid nyupptäckt hematuri/proteinuri hos en ung patient med mikroskopisk hematuri eller nedsatt njurfunktion. Finns det någon i patientens släkt som har en njursjukdom? Någon som har dialys?
Hereditär benägenhet för IgAN får betydelse när vi bedömer potentiella levande njurdonatorer inom familjen. Asymtomatisk hematuri hos potentiell levande njurdonator kan ibland accepteras, men särskild försiktighet i bedömningen måste iakttas om det gäller donator med nära släktskap med recipient som har IgAN.
Trots att IgAN har varit känd under drygt 50 år (Berger 1966) och är den dominerande orsaken till kronisk glomerulonefrit har vi fortfarande inte tillräcklig kunskap om patogenes för att kunna utveckla terapier som effektivt kan bromsa sjukdomsprogressen. En solid biomarkör med högt prediktivt värde skulle underlätta diagnostiken likaväl som bedömningen av prognos och terapival. En biomarkör skulle också vara värdefull i de fall då komplikationsrisken vid njurbiopsi bedöms som stor och där låg prognostisk riskpoäng skulle innebära att ingreppet kunde anstå. Sannolikt behövs en uppsättning av biomarkörer då patogenesen är multifaktoriell.
Viktigast i dag är att identifiera patienter med IgAN i tidig fas för att försöka fördröja progress till terminal uremi med dialys eller transplantationsbehov. Då krävs det att mikroskopisk hematuri, i synnerhet vid samtidig albuminuri, inte negligeras och framför allt inte hos unga individer. Vi vill därför rikta en uppmaning och vädjan till kollegor inom andra specialiteter att uppmärksamma dessa fynd och vid behov kontakta njurmedicinare.
När ska man misstänka IgA-nefropati?
- Ung patient med asymtomatisk mikroskopisk hematuri där mikroskopi av urinsediment visar korniga cylindrar och fyndet kvarstår vid omkontroll
- Mikroskopisk hematuri + fynd av korniga cylindrar i urinen + eventuell albuminuri
- Makroskopisk hematuri (med normalt utfall vid urologisk utredning), spontant övergående men med kvarstående mikroskopisk hematuri
- Nyupptäckt albuminuri, utan känd komorbiditet (som till exempel diabetes mellitus, generell kärlsjukdom)
- Nytillkommen kreatininstegring alternativt sänkt eGFR utan känd komorbiditet (diabetes mellitus, generell kärlsjukdom) – där pre- och postrenala orsaker till fynden har uteslutits.
Om något av ovanstående hittas, skriv remiss till njurmedicinsk enhet för utredning.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Fallbeskrivning
30-årig man inkom till akutmottagningen på grund av huvudvärk och 5 kg viktnedgång på 3 månader. Blodtryck var 200/125 mm Hg. P-kreatinin var 950 µmol/l.
Ett år tidigare hade patienten sökt vårdcentral på grund av huvudvärk och befanns då ha högt blodtryck, varför antihypertensiv behandling inleddes. På urinsticka noterades 3+ för erytrocyter, ingen albuminuri. P-kreatinin var 130 µmol/l. Ultraljudsundersökning visade normalstora njurar. Cystoskopi och urincytologi var utan anmärkning.
Vid en hälsokontroll 4 år dessförinnan hade ett gränsblodtryck (140/85 mm Hg) samt en mikroskopisk hematuri noterats, vilka lämnades utan åtgärd.
Remiss skickades från akuten till njurmottagningen för fortsatt utredning. Njurbiopsi avslöjade en IgA-nefropati med flertalet sklerotiska glomeruli samt tubulointerstitiell fibros. Patientens njursvikt progredierade ytterligare, och han påbörjade dialys kort därefter.
Referenser
- Kiryluk K, Li Y, Sanna-Cherchi S, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6):e1002765.
- Svenskt njurregister. Årsrapport 2019.
- Edström Halling S, Söderberg MP, Berg UB. Predictors of outcome in paediatric IgA nephropathy with regard to clinical and histopathological variables (Oxford classification). Nephrol Dial Transplant. 2012;27(2):715-22.
- Audemard-Verger A, Terrier B, Dechartres A, et al; French Vasculitis Study Group. Characteristics and management of IgA vasculitis (Henoch-Schönlein) in adults: data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol. 2017;69(9):1862-70.
- Lu S, Liu D, Xiao J, et al. Correlation between clinical and pathological characteristics of Henoch-Schönlein purpura nephritis in adults. Iran J Kidney Dis. 2016;11(1):12-7.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1-11.
- Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Roberts ISD, Cook HT, Troyanov S, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009;76(5):546-56.
- Trimarchi H, Barratt J, Cattran DC, et al; IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology Society. Oxford classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017;91(5):1014-21.
- Magistroni R, D’Agati VD, Appel GB, et al. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015;88(5):974-89.
- Barratt J, Rovin BH, Cattran D, et al. Why target the gut to treat IgA nephropathy? Kidney Int Rep. 2020;5(10):1620-4.
- Berthoux F, Suzuki H, Thibaudin L, et al. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol. 2012;23(9):1579-87.
- Yanagawa H, Suzuki H, Suzuki Y, et al. A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLoS One. 2014;9(5):e98081.
- Chen P, Yu G, Zhang X, et al. Plasma galactose-deficient IgA1 and C3 and CKD progression in IgA nephropathy. Clin J Am Soc Nephrol. 2019;14(10):1458-65.
- Gutiérrez E, Carvaca-Fontán F, Luzardo L, et al. A personalized update on IgA nephropathy: a new vision and new future challenges. 2020;144(11):555-71.
- Takahata A, Arai S, Hiramoto E, et al. Crucial role of AIM/CD5L in the development of glomerular inflammation in IgA nephropathy. J Am Soc Nephrol. 2020;31(9):2013-24.
- Tortajada A, Gutiérrez E, Pickering MC, et al. The role of complement in IgA nephropathy. Mol Immunol. 2019;114:123-32.
- Coppo R, Peruzzi L, Loiacono E, et al. Defective gene expression of the membrane complement inhibitor CD46 in patients with progressive immunoglobulin A nephropathy. Nephrol Dial Transplant. 2019;34(4):587-96.
- Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22(10):1795-803.
- Scolari F, Amoroso A, Savoldi S, et al. Familial clustering of IgA nephropathy: further evidence in an Italian population. Am J Kidney Dis. 1999;33(5):857-65.
- Neugut YD, Kiryluk K. Genetic determinants of IgA nephropathy: Western perspective. Semin Nephrol. 2018;5(38):443-54.
- Kiryluk K, Li Y, Scolari F, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46(11):1187-96.
- Barry MA, Simon GG, Mistry N, et al. Global trends in neglected tropical disease control and elimination: impact on child health. Arch Dis Child. 2013;98(8):635-41.
- Pullan RL, Smith JL, Jasrasaria R, et al. Global numbers of infection and disease burden of soil transmitted helminth infections in 2010. Parasit Vectors. 2014;7:37.
- Rodrigues JC, Haas M, Reich HN. IgA nephropathy. Clin J Am Soc Nephrol. 2017;12(4):677-86.
- Wheeler DC, Stefánsson BV, Jongs N, et al; DAPA-CKD Trial Committees and Investigators. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: a prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. 2021;9(1):22-31.
- Buemi M. Effect of fluvastatin on proteinuria in patients with immunoglobulin A nephropathy. Clin Pharmacol Ther. 2000;67(4):427-31.
- Pozzi C. Treatment of IgA nephropathy. J Nephrol. 2016;29(1):21-5.
- Pozzi C, Bolasco PG, Fogazzi GB, et al. Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet. 1999;353(9156):883-7.
- Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term results of a randomized, controlled trial. J Am Soc Nephrol. 2004;15(1):157-63.
- Fellström BC, Barratt J, Cook H, et al; NEFIGAN Trial Investigators. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): a double-blind, randomised, placebo-controlled phase 2b trial. Lancet. 2017;389(10084):2117-27.
- Du B, Jia Y, Zhou W, et al. Efficacy and safety of mycophenolate mofetil in patients with IgA nephropathy: an update meta-analysis. BMC Nephrol. 2017;18(1):245.
- Lundberg S, Westergren E, Smolander J, et al. B cell depleting therapy with rituximab or ofatumumab in immunoglobulin A nephropathy or vasculitis with nephritis. Clin Kidney J. 2017;10(1):20-6.
- Tumlin JA, Hennigar RA. Clinical presentation, natural history, and treatment of crescentic proliferative IgA nephropathy. Semin Nephrol. 2004;24(3):256-68.
- Cheung CK, Rajasekaran A, Barratt J, et al. An update on the current state of management and clinical trials for IgA nephropathy. J Clin Med. 2021;10(11):2493.
- Ponticelli C, Glassock RJ. Posttransplant recurrence of primary glomerulonephritis. Clin J Am Soc Nephrol. 2010;5(12):2363-72.
- Lafayette RA, Kelepouris E. Immunoglobulin A nephropathy: advances in understanding of pathogenesis and treatment. Am J Nephrol. 2018;47(Suppl 1):43-52.
- Suzuki H. Biomarkers for IgA nephropathy on the basis of multi-hit pathogenesis. Clin Exp Nephrol. 2019;23(1)26-31.
- Schena FP, Cerullo G, Rossini M, et al. Increased risk of end-stage renal disease in familial IgA nephropathy. J Am Soc Nephrol. 2002;13(2):453-60.
- Jarrick S, Lundberg S, Stephansson O, et al. Pregnancy outcomes in women with immunoglobulin A nephropathy: a nationwide population-based cohort study. J Nephrol. 2021;34(5):1591-8.
- Su X, Lv J, Liu Y, et al. Pregnancy and kidney outcomes in patients with IgA nephropathy: a cohort study. Am J Kidney Dis. 2017;70(2):262-9.
- Mahmoodpoor F, Rahbar Saadat Y, Barzegari A, et al. The impact of gut microbiota on kidney function and pathogenesis. Biomed Pharmacother. 2017;93:412-9.
- Barbour SJ, Coppo R, Zhang H, et al. Evaluating a new international risk-prediction tool in IgA nephropathy. JAMA Intern Med. 2019;179(7):942.
- Maixnerova D, Reily C, Bian Q, et al. Markers for the progression of IgA nephropathy. J Nephrol. 2016;29(4):535-41.
- Coppo R. Biomarkers and targeted new therapies for IgA nephropathy. Pediatr Nephrol. 2017;32(5):725-31.
- Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71(11):1148-54.
- Gharavi AG, Moldoveanu Z, Wyatt RJ, et al. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008;19(5):1008-14.
- Berthelot L, Robert T, Vuiblet V, et al. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015;88(4):815-22.
- Wakai K, Kawamura T, Matsuo S, et al. Risk factors for IgA nephropathy: a case-control study in Japan. Am J Kidney Dis. 1999;33(4):738-45.
- Zhang J, Ren P, Wang Y, et al. Serum matrix metalloproteinase-7 level is associated with fibrosis and renal survival in patients with IgA nephropathy. Kidney Blood Press Res. 2017;42(3):541-52.
Summary
IgA nephropathy is the most common form of inflammatory kidney disease causing uraemia world-wide and initially often a silent disease with microscopic haematuria as the only clinical finding. If left untreated, progress to terminal uraemia and dialysis is not uncommon as at least 30 % develop end stage renal failure. Awareness of the existence of the disease among GPs, internists and urologists may be helpful, not disregarding microscopic haematuria, particularly in combination with albuminuria or finding of renal casts in the urine, especially in younger individuals. No diagnostic marker in blood or urine for the disease has yet been established so kidney biopsy is still needed to confirm diagnosis. The degree of renal dysfunction, hypertension, albuminuria, and histology findings at time of diagnosis have significant impact on renal outcome. Potential biomarkers for progressive disease have been described but no one has so far been implicated in clinical practice. Until now, the only evidence-based medication consists of blockers of the renin-angiotensin-system and corticosteroids. However, new and potentially more specific drugs are under clinical investigation. Early intervention is mandatory to prevent disease progression. Thus, we want to alarm other specialists to an increased alertness for this disease, referring patients at an early stage of possible IgA nephropathy to the nephrologist for diagnosis and interventions.
1. Ravnskov U. Glomerulonefrit – en yrkessjukdom – nya rön om en sjukdom associerad med exposition för njurtoxiska ämnen. Läkartidningen 2000;97:3978-9.
2. Ravnskov U. Hydrocarbon exposure may cause glomerulonephritis and worsen renal function: evidence based on Hill´s criteria for causality. Q J Med. 2000;93:551-6. doi: 10.1093/qjmed/93.8.551
3. Hoitsma AJ, Wetzels JF, Koene RA. Drug-induced nephrotoxicity. Aetiology, clinical features and management. Drug Saf. 1991;6:131-47. doi: 10.2165/00002018-199106020-00004
4. Ravnskov U. Experimental glomerulonephritis induced by hydrocarbon exposure: A systematic review. BMC Nephrology 2005; 6:15-9. doi:10.1186/1471-2369-6-15