
Ärftlig transtyretinamyloidos är en sällsynt men livshotande sjukdom med klusterområden, bland annat i norra Sverige.
Sjukdomen kännetecknas av perifer polyneuropati, ofta i kombination med autonom neuropati, arytmier och hypertrof kardiomyopati.
Utredning, behandling och uppföljning av ärftlig transtyretinamyloidos kräver ett multidisciplinärt omhändertagande.
Tidigare kunde endast symtomlindrande behandling och levertransplantation erbjudas, men nu finns flera effektiva behandlingar, bland annat genterapi som kraftigt minskar produktionen av det sjukdomsorsakande proteinet transtyretin.
Utvecklingen av nya läkemedel har varit snabb och framgångsrik, men medför etiska och hälsoekonomiska utmaningar.
Amyloidos är ett tillstånd som kännetecknas av inlagring av olösliga proteinfibriller (amyloid) i kroppens vävnader. Sjukdomen kan vara lokaliserad eller systemisk, och både ärftliga och förvärvade former förekommer. AL-amyloidos, som orsakas av en monoklonal plasmacellssjukdom, och vildtypsformen av transtyretinamyloidos (ATTRwt-amyloidos) utgör de vanligaste förvärvade amyloidoserna. Ärftlig transtyretinamyloidos (ATTRv-amyloidos), tidigare benämnd familjär amyloidos med polyneuropati (FAP), är den vanligaste ärftliga systemiska amyloidosen och beskrevs för första gången i Sverige på 1960-talet [1]. Fram till 1990-talet fanns endast symtomlindrande behandling, och prognosen var dyster, men 1990 kom en ljusning i form av levertransplantation som i valda fall har god effekt på sjukdomsförloppet [2]. Dock fanns ett behov av farmakologiska behandlingar, och det första läkemedlet mot ATTRv-amyloidos godkändes 2011 [3]. Därefter har utvecklingen fortsatt, och de senaste 10 åren har inneburit en behandlingsmässig revolution för patienter med ATTRv-amyloidos, mycket tack vare godkännandet av genterapibehandling 2018 [3]. Denna utveckling är naturligtvis mycket positiv, men medför också etiska och hälsoekonomiska utmaningar. Syftet med vår artikel är att ge en överblick av denna sällsynta sjukdom som, med anledning av den senaste tidens behandlingsframgångar, kan ge viktiga lärdomar för framtiden.
Patofysiologi
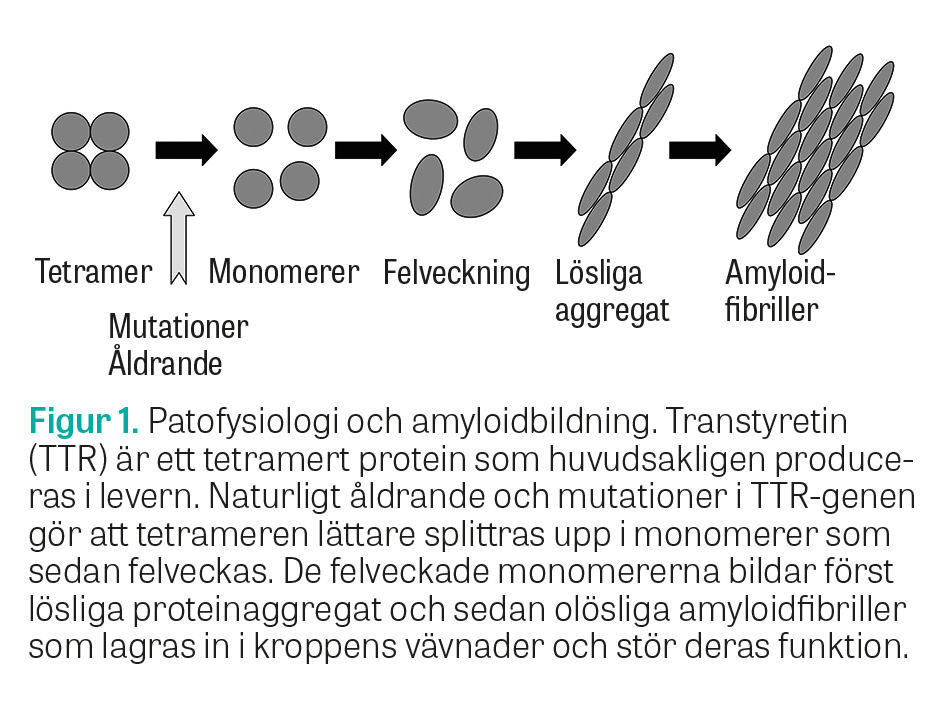
ATTRv-amyloidos är en autosomalt dominant monogen sjukdom som orsakas av mutationer i genen som kodar för proteinet transtyretin (TTR). I dag finns över 120 kända mutationer i TTR-genen [4]. Majoriteten av dessa mutationer är sjukdomsorsakande och ger en instabilitet i transtyretintetrameren, som lättare splittras upp i monomerer, som i sin tur bildar amyloid (Figur 1). Transtyretin syntetiseras främst i levern men även lokalt i ögat och i det centrala nervsystemet. Amyloidinlagringen kan i princip ske i hela kroppen, men orsakar kliniska symtom främst från det perifera nervsystemet, mag–tarmkanalen, urinvägarna, hjärtat och ögat. Sjukdomens fenotyp beror delvis på genotypen, men variationer finns också inom samma genotyp, där typen av amyloidfibriller (typ A eller B) är betydelsefull för sjukdomens fenotyp och prognos, då typ A-fibriller är associerade med amyloid kardiomyopati medan typ B-fibriller inte är det [5].
Epidemiologi
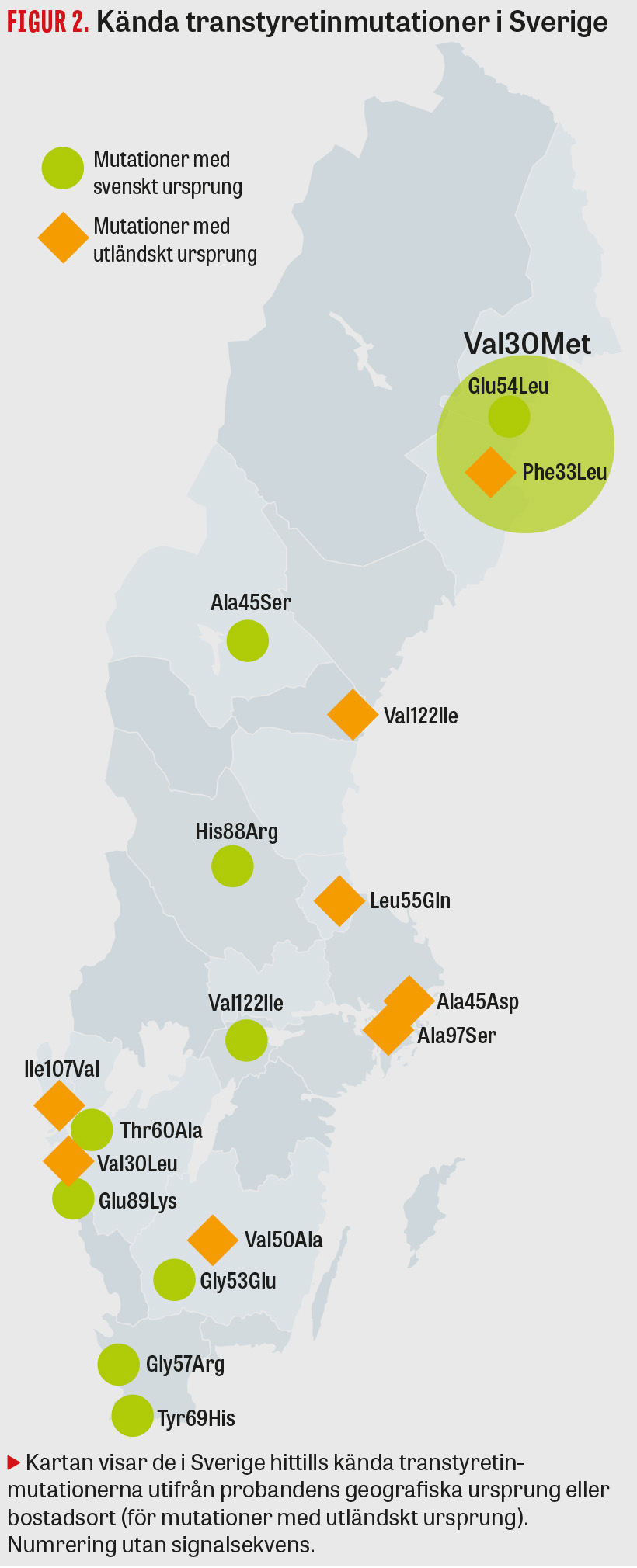
ATTRv-amyloidos är en sällsynt sjukdom som förekommer över hela världen. Den vanligaste patogena mutationen i Europa, Val30Met (p.Val50Met), är dock vanligare i vissa så kallade klusterområden i bland annat Sverige, Portugal och Japan. Globalt finns ca 10 000 patienter med sjukdomen, och den högsta prevalensen återfinns i Portugal med runt 2 000 patienter på 10 miljoner invånare [6]. Den högsta beskrivna anlagsbärarfrekvensen på 2 procent finns dock i Sverige (Västerbotten), och i dag beräknas omkring 8 000 anlagsbärare finnas i vårt land [7]. Den ofullständiga penetransen (43 procent vid 70 års ålder) [8] gör dock att antalet patienter som lever med sjukdomen i Sverige beräknas till ca 400, varav merparten (75 procent) bor i Norrbotten och Västerbotten. Ungefär 30 nya sjukdomsfall diagnostiseras årligen. En stor andel patienter och anlagsbärare härstammar från Skellefteåtrakten, varför sjukdomen ofta går under namnet Skelleftesjukan. Utöver Val30Met-mutationen, som står för 95 procent av alla sjukdomsfall i Sverige [9], finns 16 andra mutationer med svenskt eller utländskt ursprung beskrivna i landet (Tabell 1 och Figur 2) [10].
Klinisk bild
Sjukdomen kännetecknas av en sensomotorisk övervägande axonal perifer polyneuropati. Känselstörningar och/eller värk i fötter och underben utgör debutsymtom hos 90 procent av patienterna med Val30Met-mutationen. Autonom neuropati är också vanlig, och erektil dysfunktion eller störd mag–tarmfunktion kan utgöra debutsymtom. Polyneuropatin är fortskridande och ger tilltagande besvär med känselnedsättning, smärta och muskelsvaghet i ben och armar. Parallellt tilltar ofta den autonoma neuropatin med ortostatisk hypotension, hjärtarytmier, nedsatt mag–tarmmotilitet och miktionsbesvär som följd [11]. I slutstadiet av sjukdomen är patienterna rullstolsburna och undernärda, och de lider ofta av både urin- och avföringsinkontinens. Ungefär hälften av patienterna utvecklar också proteinuri eller ögonkomplikationer, oftast i form av glaskroppsgrumlingar och glaukom. En femtedel drabbas av kronisk njursvikt, och vissa behöver dialys [11]. Drygt hälften av patienterna har den amyloida fibrilltyp (typ A) som medför risk för kardiomyopati [12], och majoriteten av dessa utvecklar förr eller senare hjärtsvikt, oftast med bevarad ejektionsfraktion [13]. I vissa fall kan sjukdomen debutera med kardiella besvär [14], och kardiomyopati utgör det dominerande problemet för vissa genotyper, som TTR-mutationerna Glu54Leu, Ala45Ser, Gly53Glu och His88Arg [10]. Vid kardiomyopati är bilaterala karpaltunnelsyndrom och spinal stenos dessutom vanliga [13]. Hjärtarytmier som AV-block, sinusarrest och förmaksflimmer är också vanliga, oavsett om kardiomyopati föreligger eller inte. Omkring en av tio utvecklar pacemakerkrävande arytmier [9, 14]. Vissa genotyper medför risk för CNS-komplikationer, vilket också ses hos patienter med lång överlevnad efter levertransplantation [15]. Fynd som inger misstanke om ATTRv-amyloidos sammanfattas i Fakta 1. Medelåldern vid sjukdomsdebut är 58 år i Sverige, men debutåldern spänner mellan 22 och 84 år [9]. Utan sjukdomsmodifierande behandling är överlevnaden ca 10 år efter sjukdomsdebut [9, 11].
Diagnostik
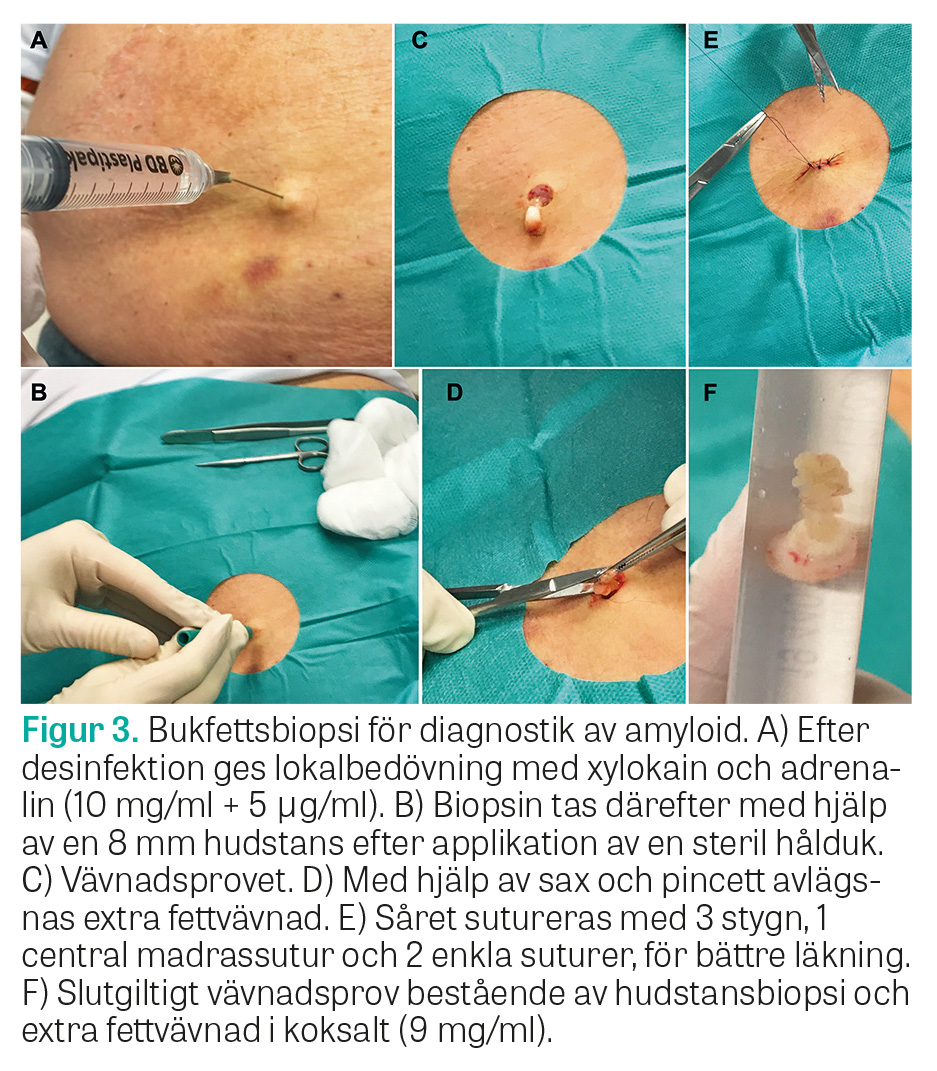
Hörnstenen i diagnostik av all amyloidos är att påvisa amyloid i vävnadsbiopsier som färgas med Kongorött och undersöks i polarisationsmikroskop. Påvisade amyloidinlagringar, sjukdomstypiska symtom och förekomst av TTR-mutation krävs för att ställa diagnosen ATTRv-amyloidos. Vid ATTRv-amyloidos är bukfettsbiopsi en träffsäker diagnostisk metod [12] som utgör förstahandsval (Figur 3), men biopsier från mag–tarmkanal, hjärta eller njure är gångbara alternativ. Vävnadsprov för amyloiddiagnostik skickas primärt till Klinisk patologi vid Akademiska sjukhuset i Uppsala, som specialiserat sig på denna diagnostik. DNA-analys av TTR-genen görs vid de kliniskt genetiska enheterna vid Sveriges universitetssjukhus och ska föregås av genetisk vägledning. Vid misstänkt ATTR-amyloidos bör DNA-analys utföras frikostigt, då ATTRwt-amyloidos är svår att skilja från ATTRv-amyloidos av typ A. Vid misstanke om amyloid kardiomyopati kan skintigrafi med teknetium-99m-märkt DPD (butedronsyra) ersätta vävnadsprov för diagnostik, så länge AL-amyloidos har uteslutits [16]. Vid minsta tveksamhet ska dock vävnadsbiopsi tas, då bilden vid ATTR- och AL-amyloidos kan vara snarlik [17]. Utöver detta bör patienterna undersökas utifrån klinisk bild med exempelvis neurofysiologiska test, EKG, långtids-EKG, ekokardiografi, MR hjärta, endoskopi, urinprov och blodprov för att diagnostisera eventuella komplikationer.
Behandling
Utöver symtomlindrande behandling finns i dag flera effektiva sjukdomsmodifierande terapier. Sedan det första läkemedlet mot ATTRv-amyloidos godkändes 2011 har utvecklingen av nya läkemedel varit snabb och framgångsrik. Godkännandet av genterapi mot ATTRv-amyloidos listades av tidskriften Science som ett av de 10 viktigaste vetenskapliga genombrotten under 2018. Nedan beskrivs de behandlingar som är tillgängliga i dag i kronologisk ordning.
Levertransplantation. Den första levertransplantationen vid ATTRv-amyloidos genomfördes i Sverige 1990. Hypotesen var att man genom att byta ut levern skulle häva dess produktion av muterat transtyretin och därmed bromsa sjukdomsförloppet, i praktiken en form av genterapi. De tidiga resultaten var lovande [18], vilket naturligtvis var ett stort framsteg i behandlingen av denna dödliga sjukdom vars symtom tidigare bara kunde lindras. Efter hand blev det dock tydligt att alla patienter inte hade samma goda effekt av transplantation, och lång sjukdomsduration och malnutrition har visat sig vara prediktorer för sämre överlevnad [2]. Överlevnaden efter transplantation är också kortare för patienter med andra mutationer än Val30Met samt för dem med sjukdomsdebut efter 50 års ålder, vilket kan kopplas till den amyloida fibrilltypen (typ A) och en fortsatt vävnadsinlagring av vildtyps-TTR efter transplantationen [5]. Bäst prognos efter levertransplantation har patienter med sjukdomsdebut före 50 års ålder och Val30Met-mutationen, för vilka 15-årsöverlevnaden är nära 80 procent [2]. Totalt har 150 patienter med ATTRv-amyloidos levertransplanterats i Sverige [9], varav 83 har genomgått så kallad dominotransplantation, då den uttagna levern (som bortsett från produktion av variant transtyretin fungerar normalt) donerats vidare till en patient med svår leversjukdom. Ett fåtal patienter har även genomgått hjärt- eller njurtransplantation [9]. Antalet levertransplantationer på denna indikation har minskat kraftigt de senaste åren tack vare tillkomsten av farmakologiska behandlingar [2]. Med anledning av detta, och att vissa patienter utvecklat (de novo) amyloidos redan 5–10 år efter att de mottagit en lever med TTR-mutation, har även antalet dominotransplantationer minskat betydligt.
Tafamidis. Tafamidis stabiliserar transtyretintetrameren, vilket motverkar amyloidbildning och bromsar sjukdomsförloppet hos patienter med polyneuropati i tidigt skede [19]. Läkemedlet (i dosen 20 mg) godkändes i EU 2011 för behandling av ATTRv-amyloidos med polyneuropati i stadium I (inte i behov av gånghjälpmedel). Tafamidis 20 mg är inte förmånsberättigat i Sverige. Långtidsdata visar att tafamidis tolereras väl och indikerar överlevnadsvinster, även om neuropatin tenderar att progrediera över tid [3]. Som vid levertransplantation svarar patienter med Val30Met-mutationen i tidigt sjukdomsskede bäst på behandlingen. En knapp tredjedel av patienterna har dock ingen synbar effekt av tafamidis, vilket delvis kan korreleras till manligt kön och lägre transtyretinkoncentration i plasma vid behandlingsstart [3]. Tafamidis har senare också utvärderats avseende effekt på amyloid kardiomyopati, då i dosen 61 mg, och har visat sig reducera mortalitet och hjärtrelaterad sjukhusvård vid ATTRv- och ATTRwt-amyloidos med kardiomyopati [20]. Tafamidis 61 mg godkändes i EU 2020 för behandling av ATTR-kardiomyopati och blev 2021 förmånsberättigat i Sverige för behandling av patienter med dominerande kardiomyopati.
Diflunisal. Liksom tafamidis stabiliserar diflunisal transtyretintetrameren och motverkar därmed amyloidbildning. Diflunisal (500 mg dagligen) har i en randomiserad studie visats kunna bromsa sjukdomsförloppet vid ATTRv-amyloidos med polyneuropati oberoende av genotyp och svårighetsgrad [21]. Mindre öppna behandlingsstudier indikerar också en stabiliserande effekt på ATTR-kardiomyopati [3,13]. Eftersom diflunisal är ett NSAID-preparat finns viss risk för biverkningar som gastrit, mag–tarmblödning och njurpåverkan. Diflunisal är inte godkänt för behandling av ATTRv-amyloidos, utan kan endast förskrivas på licens till patienter som inte kan behandlas med godkända läkemedel.
Patisiran. Patisiran är en liten dubbelsträngad ribonukleinsyra (RNA) som minskar produktionen av transtyretin i hepatocyterna genom RNA-interferens och nedbrytning av TTR-mRNA. Detta leder till en reduktion av både variant och vildtyps-TTR. Patisiran givet som en intravenös infusion 0,3 mg/kg var tredje vecka har i en randomiserad studie visat sig minska transtyretin i serum med i genomsnitt 81 procent och effektivt stoppa progress av polyneuropati oberoende av genotyp och utan allvarligare biverkningar [22]. Subgruppsanalyser antyder också en bromsande effekt på ATTR-kardiomyopati. Läkemedlet godkändes i EU 2018 för behandling av ATTRv-amyloidos med polyneuropati i tidigt och medelsvårt skede (stadium I och II). För att motverka infusionsrelaterade biverkningar ges premedicinering med kortison, paracetamol samt histaminreceptorblockerare. Dagligt tillskott av vitamin A rekommenderas under pågående behandling för att motverka vitamin A-brist sekundärt till lägre nivåer av transtyretin i serum. Patisiran är under utökad övervakning, men hittills har inga nya eller allvarliga säkerhetsrisker framkommit. Läkemedlet är inte förmånsberättigat i Sverige.
Inotersen. Inotersen är en enkelsträngad antisensoligonukleotid som minskar produktionen av transtyretin i hepatocyterna genom att inducera nedbrytning av TTR-mRNA. Detta leder till en reduktion av både variant och vildtyps-TTR. Inotersen givet som en subkutan injektion 284 mg varje vecka har i en randomiserad studie visat sig minska transtyretin i serum med i genomsnitt 74 procent och effektivt stoppa progress av polyneuropati oberoende av genotyp [23]. En öppen behandlingsstudie har också indikerat positiv effekt på ATTR-kardiomyopati [3]. Fall av glomerulonefrit och trombocytopeni har dock rapporterats, varav ett fall med dödlig utgång på grund av intrakraniell blödning och samtidig trombocytopeni. Läkemedlet godkändes i EU 2018 för behandling av ATTRv-amyloidos med polyneuropati i tidigt och medelsvårt skede (stadium I och II). Ingen premedicinering är nödvändig; dock rekommenderas kontroll av trombocyter åtminstone varannan vecka samt övervakning av njur- och leverfunktion. Dagligt tillskott av vitamin A rekommenderas under pågående behandling för att motverka vitamin A-brist sekundärt till lägre nivåer av transtyretin i serum. Inotersen är under utökad övervakning, men hittills har inga nya säkerhetsrisker framkommit. Läkemedlet är inte förmånsberättigat i Sverige.
Uppföljning
Eftersom ATTRv-amyloidos är en systemisk sjukdom krävs ett multidisciplinärt omhändertagande av patienterna. Behovet av specialistkompetens varierar med den kliniska bilden samt under sjukdomsförloppet, men neurolog, kardiolog, gastroenterolog, oftalmolog och nefrolog behöver vanligen konsulteras. Konsultation av klinisk genetiker är också värdefull, särskilt vid familjescreening, vilket bör övervägas för vissa genotyper och för släkter med ett mer aggressivt sjukdomsförlopp. Klinisk uppföljning sker med fördel halvårsvis av specialist i internmedicin, neurologi, gastroenterologi eller kardiologi med klinisk bedömning, blodprov, urinprov och EKG. Långtids-EKG bör utföras årligen för att fånga eventuella behandlingskrävande arytmier, och vid påvisad kardiomyopati bör ekokardiografi genomföras åtminstone vartannat år. Utvärdering av effekten av sjukdomsmodifierande läkemedel görs enligt NT-rådets rekommendationer i samband med klinisk uppföljning [24, 25]. Ett nationellt kvalitetsregister (Svenska transtyretinamyloidosregistret, SveATTR) har upprättats för att underlätta klinisk uppföljning och forskning kring ATTR-amyloidos [9], och nyupptäckta fall bör rapporteras till registret.
Hälsoekonomi
Fram till 2018 var kostnaderna för behandling av ATTRv-amyloidos hanterbara. Före 2011 utfördes i första hand levertransplantationer, och mellan 2011 och 2018 användes diflunisal och tafamidis 20 mg parallellt med levertransplantation. En levertransplantation medför en engångskostnad på omkring 700 000 kr, medan diflunisal kostar drygt 10 000 kr per patient och år och marknadspriset för tafamidis 20 mg är cirka 1,2 miljoner kr per patient och år. Sedan 2018 har förskrivningen av diflunisal minskat till förmån för behandling med tafamidis, patisiran och inotersen. Patisiran har en årskostnad på 3,7–4,4 miljoner kr per patient och inotersen en årskostnad på ca 3,1 miljoner kr per patient. Efter godkännandet av tafamidis 61 mg mot ATTR-kardiomyopati 2020 kan diflunisal i praktiken endast förskrivas till patienter i slutskedet av sjukdomen (stadium III), trots att tafamidis 61 mg har en årskostnad på ca 1,3 miljoner kr per patient och endast har begränsad subvention. En analys från USA anger att priset på tafamidis 61 mg behöver sänkas med 93 procent för att det ska bli kostnadseffektivt [26]. Även om de nationella avtal som tecknats för tafamidis, patisiran och inotersen ger viss ekonomisk återbäring till regionerna så medför ovanstående utveckling en mycket kraftig ökning av läkemedelskostnaderna, särskilt i Norrbotten och Västerbotten. Detta medför i sin tur etiska utmaningar eftersom alla patienter inte får samma tillgång till de mest (kostnads)-effektiva behandlingarna. NT-rådets rekommendationer [24, 25] är därför ett viktigt stöd vid behandlingsval och utvärdering av behandlingseffekt.
Klinisk forskning
Trots att 3 läkemedel redan är godkända för behandling av ATTRv-amyloidos pågår flera kliniska läkemedelsprövningar runtom i världen [3]. AG10 och tolkapon utgör nya potentiella transtyretinstabiliserande läkemedel, varav det senare även passerar blod–hjärnbarriären, vilket gör att det förhoppningsvis även kan motverka CNS-komplikationer. Vutrisiran och eplontersen, som är vidareutvecklingar av patisiran respektive inotersen, utgör båda lovande subkutana alternativ för att minska transtyretinproduktionen i levern via nedbrytning av mRNA. Även monoklonala antikroppar mot TTR-amyloid är under klinisk prövning [3]. Mest intressanta är dock de tidiga resultaten från en pågående studie av NTLA-2001, som via en dos med Crispr/Cas9-baserad genterapi permanent skulle kunna minska transtyretin i serum hos patienter med ATTRv-amyloidos utan allvarligare biverkningar [27].
Konklusion
Tack vare det senaste decenniets revolutionerande utveckling finns i dag flera behandlingar som effektivt hindrar progressen av denna sällsynta men livshotande sjukdom. Behandlingen måste dock påbörjas i tidigt skede för bästa effekt, vilket kräver en snabb och korrekt diagnostik. Så länge inga nya säkerhetsrisker framkommer ser prognosen i dag mycket god ut för patienter som behandlas med genterapi, och behandling med Crispr/Cas9-teknik ser ut att kunna bli nästa stora framgång i behandlingen av ATTR-amyloidos. På sikt kan även kombinationsbehandling bli aktuell för att motverka CNS-komplikationer. Hälsoekonomiskt finns dock stora utmaningar eftersom de nya terapierna är dyra och medför skenande läkemedelskostnader. Framtiden måste utvisa hur dessa kostnader bäst fördelas regionalt och nationellt så att alla patienter kan få tillgång till den mest effektiva behandlingen.
Läs även författarintervjun med Jonas Wixner.
Potentiella bindningar eller jävsförhållanden: Jonas Wixner är huvudprövare för två kliniska läkemedelsprövningar och medprövare för en klinisk läkemedelsprövning sponsrade av Alnylam Pharmaceuticals, huvudprövare för en klinisk läkemedelsprövning sponsrad av Akcea Therapeutics och medprövare för en klinisk läkemedelsprövning sponsrad av Intellia Therapeutics, har tidigare varit svensk huvudansvarig för en internationell forskningsdatabas (THAOS) sponsrad av Pfizer samt har erhållit arvoden till arbetsgivaren från Pfizer, Alnylam Pharmaceuticals och Akcea Therapeutics för föreläsningsuppdrag och deltagande i rådgivande kommittéer. Intissar Anan är medprövare för kliniska läkemedelsprövningar sponsrade av Alnylam Pharmaceuticals, Akcea Therapeutics, Intellia Therapeutics och Prothena Corporation. Björn Pilebro är huvudprövare för en klinisk läkemedelsprövning och medprövare för en klinisk läkemedelsprövning sponsrade av Alnylam Pharmaceuticals, medprövare för en klinisk läkemedelsprövning sponsrad av Akcea Therapeutics och huvudprövare för en läkemedelsprövning sponsrad av Intellia Therapeutics samt har erhållit arvoden till arbetsgivaren från Pfizer och Alnylam Pharmaceuticals för föreläsningsuppdrag. Erica Uneus är medprövare för kliniska läkemedelsprövningar sponsrade av Alnylam Pharmaceuticals, Akcea Therapeutics och Prothena Corporation. Jorge Mejia Baranda har erhållit ersättning från Pfizer för medverkan på vetenskaplig kongress. Ole B Suhr är huvudprövare och medprövare för läkemedelsstudier sponsrade av Alnylam Pharmaceuticals, Intellia Therapeutics och Prothena Corporation samt har erhållit arvoden till arbetsgivaren från Pfizer, Akcea Therapeutics, Intellia Therapeutics och Alnylam Pharmaceuticals för föreläsningsuppdrag och deltagande i rådgivande kommittéer.
Fakta 1. Fynd som enskilt eller tillsammans inger misstanke om ATTRv-amyloidos
Kliniska fynd
- Progressiv perifer polyneuropati
- Autonom neuropati (inklusive mag–tarmsymtom)
- Ofrivillig viktnedgång
- Ärftlighet (hälften av fallen)
- Ursprung från Norrbotten eller Västerbotten
- Hjärtsvikt (oftast efter 50 års ålder)
- Arytmier (AV-block, sinusarrest, förmaksflimmer)
- Synrubbning (glaskroppsgrumling, glaukom)
- Proteinuri och njursvikt
Neurofysiologiska fynd
- Patologisk temperatursinnestestning
- Sensomotorisk axonal polyneuropati med distal övervikt
Ekokardiografiska fynd
- Hjärtmuskelhypertrofi
- Diastolisk dysfunktion
- »Apical sparing« i vänster kammare vid strain-analys
Fynd vid skelettskintigrafi
- Ökat kardiellt isotopupptag
(uppdaterad 2022-06-07)
Referenser
- Andersson R, Kassman T. Vitreous opacities in primary familial amyloidosis. Acta Ophthalmol (Copenh). 1968;46(3):441-7.
- Ericzon BG, Wilczek HE, Larsson M, et al. Liv-er transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847-54.
- Russo M, Gentile L, Toscano A, et al. Advances in treatment of ATTRv amyloidosis: state of the art and future prospects. Brain Sci. 2020;10(12):E952.
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35(9):E2403-12.
- Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281(4):337-47.
- Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin famil-ial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829-37.
- Olsson M, Jonasson J, Cederquist K, et al. Frequency of the transthyretin Val30Met mutation in the northern Swedish population. Amyloid. 2014;21(1):18-20.
- Gorram F, Olsson M, Alarcon F, et al. New data on the genetic profile and penetrance of hereditary Val30Met transthyretin amyloidosis in Sweden. Amyloid. 2021;28(2):84-90.
- Svenska transtyretinamyloidosregistret (SveATTR). Årsrapport 2020. https://regionvasterbotten.se/sveattr
- Suhr OB, Wixner J, Pilebro B, et al. The Swedish landscape of hereditary ATTR amyloidosis. Amyloid. 2017;24(sup1):93-4.
- Andersson R. Familial amyloidosis with polyneuropathy. A clinical study based on patients living in northern Sweden. Acta Med Scand Suppl. 1976;590:1-64.
- Paulsson Rokke H, Sadat Gousheh N, Westermark P, et al. Abdom-inal fat pad biopsies exhibit good diagnostic accuracy in patients with suspected transthyretin amyloidosis. Orphanet J Rare Dis. 2020;15(1):278.
- Linnér E, Pilebro B, Eldhagen P, et al. Hjärtamyloidos – nya möjligheter vid sjukdom med dålig prognos. Läkartidningen. 2021;118:20138.
- Arvidsson S, Pilebro B, Westermark P, et al. Amyloid cardiomyop-athy in hereditary transthyretin V30M amyloidosis – impact of sex and amyloid fibril composition. PLoS One. 2015;10(11):e0143456.
- Wange N, Anan I, Ericzon BG, et al. Atrial fibrillation and central nervous complications in liver transplanted hereditary transthyretin amyloidosis patients. Transplantation. 2018;102(2):e59-66.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404-12.
- Quarta CC, Zheng J, Hutt D, et al. 99mTc-DPD scintigraphy in immunoglobulin light chain (AL) cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2021;22(11):1304-11.
- Holmgren G, Ericzon BG, Groth CG, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113-6.
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropa-thy: a randomized, controlled trial. Neurology. 2012;79(8):785-92.
- Maurer MS, Schwartz JH, Gundapaneni B, et al; ATTR-ACT Study Investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007-16.
- Berk JL, Suhr OB, Obici L, et al; Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneurop-athy: a randomized clinical trial. JAMA. 2013;310(24):2658-67.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for her-editary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11-21.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22-31.
- NT-rådets expertgrupp för transtyretinamyloidos. Tegsedi (inotersen) och Onpattro (patisiran) vid ärftlig transtyretinamyloidos (ATTRv-amyloidos, Skelleftesjukan). NT-rådets yttrande till regionerna 2021-09-03 [citerat 6 sep 2021]. https://janusinfo.se/nationelltinforandeavlakemedel/produktinfo/onpattropatisiran
- NT-rådets expertgrupp för transtyretinamyloidos. Vyndaqel 61 mg (tafamidis) vid vildtyp (ATTRwt) eller ärftlig (ATTRv) transtyretinamyloidos hos vuxna med kardiomyopati. NT-rådets yttrande till regionerna 2021-09-03 [citerat 6 sep 2021]. https://janusinfo.se/nationelltinforandeavlakemedel/produktinfo/vyndaqeltafamidis
- Kazi DS, Bellows BK, Baron SJ, et al. Cost-effectiveness of tafamidis therapy for transthyretin amyloid cardiomyop-athy. Circulation. 2020;141(15):1214-24.
- Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene edit-ing for transthyretin amyloidosis. N Engl J Med. 2021;385(6):493-502.
Summary
Hereditary transthyretin (ATTRv) amyloidosis is a rare but life-threatening multi-systemic disease with clustering areas in, for example, northern Sweden. Until the 1990s, only symptomatic treatments were available but liver transplantation has, in selected patients, been a good therapeutic option since. The first disease-modifying drug for ATTRv amyloidosis was approved in 2011 and since then, the development of new therapeutic drugs has been rapid and successful. Two gene silencing therapies were approved for the disease in 2018, both showing a robust reduction in serum transthyretin levels and a satisfactory safety profile. Recently, CRISPR-Cas9 gene editing has also shown promising results in patients with ATTRv amyloidosis. The recent developments have had a paramount effect on the management of these patients, and will probably also have a significant positive effect on their life expectancy. However, treatment costs have skyrocketed, which implies future challenges.