
Interstitiell lungsjukdom kan vara första tecken på en autoimmun systemsjukdom. Vid myositassocierad interstitiell lungsjukdom kan lungsjukdomen vara enda symtom, utan förekomst av muskelsvaghet eller andra, extramuskulära manifestationer.
Antinukleära antikroppar kan vara negativa. Testning för myositantikroppar bör övervägas som ett led i diagnostiken vid välgrundad klinisk misstanke om interstitiell lungsjukdom av oklar genes.
Vid misstanke om interstitiell lungsjukdom bör dessa patienter remitteras till specialistklinik för utredning och om möjligt diskuteras på multidisciplinär konferens.
Tidig misstanke om inflammatorisk systemsjukdom, snabb upptäckt och tidigt insatt behandling är helt avgörande för framtida prognos, livskvalitet och överlevnad.
Interstitiell lungsjukdom kan vara den första och ibland enda manifestationen av inflammatorisk systemsjukdom. Interstitiell lungsjukdom kan förekomma vid en rad olika systemsjukdomar, såsom reumatoid artrit, systemisk skleros och myosit. Patienter med myosit kan insjukna med interstitiell lungsjukdom som första symtom, helt i avsaknad av muskelsvaghet och andra extramuskulära manifestationer, men med så kallade myositspecifika autoantikroppar som är typiska för sjukdomen. Syftet med den här artikeln är att öka medvetenheten om detta tillstånd och om värdet av serologisk testning av myositspecifika och myositassocierade autoantikroppar i diagnostiken för att kunna initiera immunmodulerande behandling.
Interstitiell lungsjukdom
Interstitiell lungsjukdom är en heterogen grupp av lungsjukdomar som karakteriseras av inflammation och fibros i lungparenkymet. Tillståndet kallas i engelskspråkig litteratur för »diffuse parenchymal lung disease« och ger vanligen en restriktiv lungfunktionsnedsättning. Vissa former av interstitiell lungsjukdom, som till exempel organiserande pneumoni, har bättre prognos och svarar på immunmodulerande behandling, medan idiopatisk lungfibros inte förbättras av sådan behandling utan är förenad med en hög dödlighet och en femårsöverlevnad på 20–40 procent [1, 2].
Interstitiell lungsjukdom vid reumatiska sjukdomar är en viktig bidragande orsak till ökad morbiditet och mortalitet. Prevalensen är mycket varierande, från 10–30 procent hos patienter med reumatoid artrit, primärt Sjögrens syndrom eller systemisk lupus erythematosus till närmare 90 procent hos patienter med myosit, systemisk skleros eller blandad bindvävssjukdom (mixed connective tissue disease) [3]. Jämfört med idiopatisk lungfibros insjuknar patienter som har reumatisk sjukdom med interstitiell lungsjukdom vid lägre ålder, och icke-rökande kvinnor är överrepresenterade [3, 4]. Förutom patienter med underliggande definierad reumatisk sjukdom bedöms ytterligare 10–20 procent av patienter med interstitiell lungsjukdom ha en oklar inflammatorisk systemsjukdom baserat på kliniska fynd och serologi utan att uppfylla kriterierna för en specifik reumatisk sjukdom [5, 6]. Tillståndet kallas då för »interstitiell pneumoni med autoimmuna kännetecken«. Eftersom lungsjukdomen kan debutera före reumatisk sjukdom eller innan denna diagnos har ställts kan prevalensen vara underskattad. Interstitiell lungsjukdom associerad med reumatisk sjukdom bör misstänkas vid förändringar på högupplösande datortomografi (HRCT) hos patienter med systemsjukdom eller hos yngre patienter, företrädesvis kvinnor, där annan genes till interstitiell lungsjukdom har uteslutits. I denna översikt fokuserar vi på interstitiell lungsjukdom associerad med myositantikroppar.
Myosit
Idiopatiska inflammatoriska myopatier är en grupp sällsynta sjukdomar som tillsammans kallas för myositer. Prevalensen är cirka 14/100 000 individer både i Sverige och i världen. Den årliga incidensen i Sverige ligger på cirka 11 fall per 1 miljon invånare, med en högre förekomst hos personer i åldern 50–79 år [7, 8]. Interstitiell lungsjukdom förekommer hos vuxna med myosit i alla åldrar, i upp till 65 procent av fallen redan vid diagnos, medan det hos barn med juvenil dermatomyosit är ovanligt. Morbiditeten är hög och botande behandling saknas. Den typiska symtombilden vid myosit består av symmetrisk proximal svaghet i skelettmuskulatur i kombination med extramuskulära symtom, som kan involvera lungor, hjärta, hud, leder och mag–tarmkanalen. De extramuskulära symtomen kan bland annat yttra sig som dyspné och torrhosta sekundärt till interstitiell lungsjukdom, Raynauds fenomen, hudmanifestationer, feber och artrit. Symtombilden är dock mycket heterogen.
Under senare år har det skett genombrott som ger stöd för diagnostiken. Man har lyckats identifiera flera autoantikroppar som förekommer vid myositassocierad interstitiell lungsjukdom, dels myositspecifika autoantikroppar och dels myositassocierade autoantikroppar, som också kan förekomma vid andra inflammatoriska systemsjukdomar. Särskilt de myositspecifika autoantikropparna har visat sig vara mycket användbara för att identifiera patienter med myosit och dessutom särskilja homogena kliniska fenotyper, varav några med stark association till interstitiell lungsjukdom (Tabell 1) [9-11].
Interstitiell lungsjukdom med myositautoantikroppar
En nyligen identifierad subgrupp av patienter med myosit insjuknar med interstitiell lungsjukdom som första sjukdomsmanifestation helt i avsaknad av muskulära symtom eller andra, extramuskulära manifestationer. Det som skiljer dessa patienter från övriga med interstitiell lungsjukdom är att de bär på myositspecifika autoantikroppar. Diagnos och behandling fördröjs ofta eftersom kliniska tecken på systemsjukdom saknas. Behandling med immunmodulerande läkemedel bidrar ofta till förbättring av de subjektiva lungsymtomen och objektiv förbättring av lungfunktion och HRCT-förändringar. Obehandlad sjukdom har en allvarlig prognos med hög morbiditet och mortalitet [12].
Utredning av interstitiell lungsjukdom
Diagnosen interstitiell lungsjukdom ställs oftast baserat på klinisk bild, radiologi och lungfunktionstest, kompletterat med resultat från bronkoskopi och ibland histopatologi. Ofta är diagnostiken komplicerad och kräver diskussion på multidisciplinär konferens.
Radiologi. Lungröntgen är ofta första steget vid utredning av symtom från lungan. Tyvärr har lungröntgen låg sensitivitet för diagnostik av interstitiell lungsjukdom, och om misstanke finns bör HRCT lungor erbjudas. Förändringar på HRCT lungor kan förekomma utan att patienten har hunnit utveckla kliniska symtom eller lungfunktionsnedsättning. Icke-specifik interstitiell pneumoni (non-specific interstitial pneumonia, NSIP) är det vanligast förekommande mönstret på HRCT vid interstitiell lungsjukdom associerad med reumatisk sjukdom. Dock kan även en bild som vid vanlig interstitiell pneumoni (usual interstitial pneumonia, UIP), organiserad pneumoni (OP) och diffus alveolär skada (diffuse alveolar damage, DAD) förekomma [13].
Bronkoskopi och histopatologi. Bronkoskopi med provtagning är värdefull för att utesluta malignitet och infektion. Vid interstitiell lungsjukdom kan cellfördelning i bronkoalveolärt lavage vara vägledande vid diagnostik, och en generell bedömning av grad av inflammation eller abnormal cellfördelning, som eosinofili, kan erhållas. Det finns emellertid ingen specifik cellfördelning som är typisk för interstitiell lungsjukdom associerad med myosit. Transbronkiella lungbiopsier eller kryobiopsier kan utföras vid bronkoskopi och kan ge provmaterial för diagnostik från perifer lungvävnad. Kirurgisk lungbiopsi, ofta med videoassisterad teknik, kan ibland bli nödvändig för definitiv histopatologisk diagnos.
Lungfunktionstest med spirometri. Interstitiell lungsjukdom ger vanligen en restriktiv lungfunktionsnedsättning med sänkt total lungkapacitet (TLC). Det är viktigt att dessa patienter genomgår en fullständig spirometri inkluderande diffusionskapacitetsbestämning (DLCO). Diffusionskapacitet är ett känsligt test som tidigt kan indikera diffusionshinder. Forcerad vitalkapacitet (FVC) kan vara sänkt både vid obstruktiv lungsjukdom och vid interstitiell lungsjukdom. Sex minuters gångtest eller arbetsprov med saturationsmätning kan också fånga diffusionshinder. Dock kan interstitiell lungsjukdom förekomma med normal lungfunktion.
Serologi. På alla svenska universitetssjukhus finns i dag en panel med 14 myositautoantikroppar som kan beställas som ett steg i utredningen av myositassocierad interstitiell lungsjukdom.
I en vanlig ANA-undersökning, som mäter förekomsten av antinukleära antikroppar, ingår endast en myositspecifik autoantikropp, anti-Jo-1, samt oftast en myositassocierad autoantikropp, anti-SSA/Ro52. Övriga autoantikroppar som kan påvisas vid myosit måste beställas separat. ANA är positiva hos 50–80 procent av patienter med myosit, men negativa ANA utesluter inte förekomst av myosit [11] eftersom de flesta myositspecifika autoantikroppar är riktade mot cytoplasmaproteiner i cellerna och kan medföra ett negativt svar [14]. Fynd av cytoplasmatiskt mönster vid ANA-undersökning kan inge misstanke om förekomst av myositspecifika autoantikroppar, men förekomsten rapporteras inte alltid av laboratoriet då en konventionell ANA-undersökning, det vill säga infärgning av cellkärnan, i sådana fall ofta saknas [15, 16].
Myositspecifika och myositassocierade autoantikroppar
Flera autoantikroppar som förekommer vid myosit har på senare år kunnat kopplas till interstitiell lungsjukdom. Dit hör gruppen antisyntetas-autoantikroppar, där anti-Jo-1 är den vanligaste. Dessa autoantikroppar förknippas med antisyntetassyndrom, som består av en kombination av de kliniska manifestationerna av interstitiell lungsjukdom, myosit, »mekanikerhänder« (hyperkeratotiska hudförändringar på sidorna av fingrarna), Raynauds fenomen och artrit. 65–100 procent av patienter med dessa autoantikroppar har tecken på interstitiell lungsjukdom, även om kliniska symtom på lungpåverkan kan saknas. I en studie med anti-Jo-1-positiva patienter debuterade 32 procent med isolerad interstitiell lungsjukdom [17-19]. Andra antisyntetas-autoantikroppar som associeras med interstitiell lungsjukdom är anti-PL-7, anti-PL-12, anti-EJ och anti-OJ. En annan myositspecifik autoantikropp med stark association till interstitiell lungsjukdom är anti-MDA-5. Av dessa autoantikroppar är anti-MDA-5, anti-PL-7 och anti-PL-12 associerade med en högre dödlighet jämfört med anti-Jo-1 [20, 21]. Antisyntetas-autoantikroppar förekommer ofta tillsammans med anti-SSA/Ro52, som i detta fall inte ska inge misstanke om SLE eller Sjögrens syndrom. I stället innebär detta att risken för interstitiell lungsjukdom ökar, jämfört med om man påvisat isolerade antisyntetas-autoantikroppar.
Anti-MDA-5 är ofta associerad med en snabbt förlöpande interstitiell lungsjukdom av typen DAD, med hög mortalitet. Dessa patienter har vanligen hudförändringar, som vid dermatomyosit, men kan sakna muskelsymtom, och tillståndet kallas då amyopatisk dermatomyosit. Hos asiater med dermatomyosit och samtidig förekomst av anti-MDA-5 föreligger en risk på upp till 80 procent att utveckla snabbt progredierande interstitiell lungsjukdom med hög mortalitet [22]. Förekomst av anti-MDA-5 och samtidigt lungengagemang ska alltid betraktas som allvarligt, och dessa patienter kan snabbt försämras. Kraftfull immunmodulerande behandling är motiverad redan från början [23].
Övriga autoantikroppar som har diagnostiskt värde vid utredning av mysoit associerad med interstitiell lungsjukdom är anti-PM-Scl och anti-Ku [24]. Bland patienter med myosit och antikropparna anti-PM-Scl eller anti-Ku förekommer interstitiell lungsjukdom hos 38 procent respektive 27 procent [17, 25].
Behandling
Val av behandling baseras på klinisk fenotyp, autoantikroppsprofil, demografi och grad av lungpåverkan [26, 27]. Vid myositassocierad interstitiell lungsjukdom ges immunmodulerande behandling i form av en initialt hög dos av kortison, ofta i form av intravenös pulsbehandling, i kombination med cyklofosfamid, mykofenolatmofetil, takrolimus eller rituximab. Vid progredierande fibrotisk interstitiell lungsjukdom kan behandling med nintedanib bli aktuell. Vid terapisvikt kan kombinationsbehandling med fler läkemedel ges [28]. Multidisciplinära konferenser med lungläkare, reumatologer och radiologer är av stort värde.
Fallbeskrivningar
Vi beskriver här tre patienter, där myositsjukdom debuterade med interstitiell lungsjukdom som enda symtom och serologisk testning var avgörande för diagnostiken.
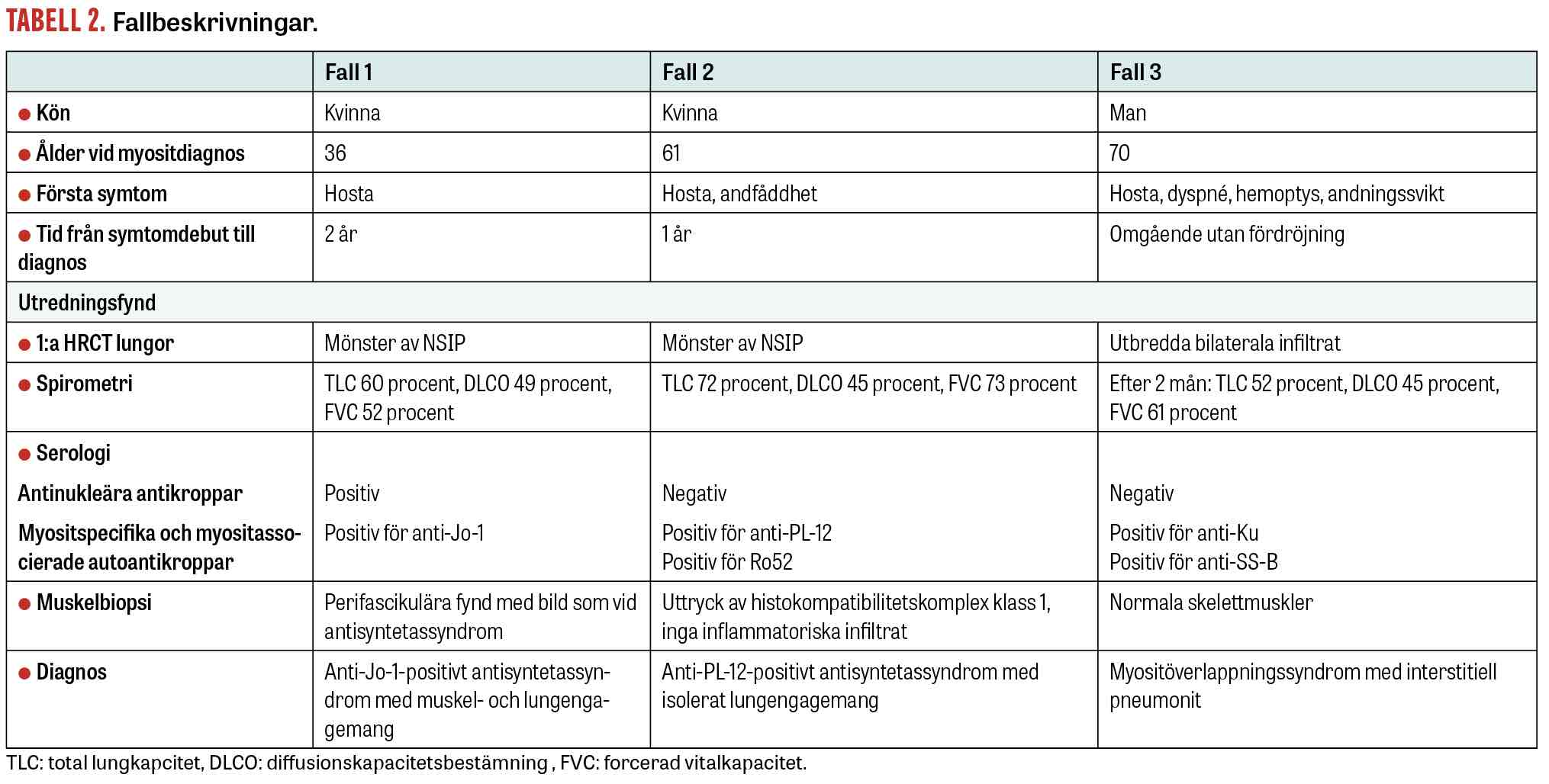
Fall 1. Den första patienten är en drygt 30-årig icke-rökande och tidigare väsentligen frisk kvinna, som insjuknade med torrhosta utan feber eller CRP-stegring. Hon bedömdes initialt ha en viros. En lungröntgen tolkades som stasbild. Patienten hade fortsatt hosta, men på grund av graviditet utfördes ingen ny radiologi. Två år senare tillkom muskelsvaghet, ökad trötthet och värk i ben och händer. Misstanke om myosit väcktes, och utredning på vårdcentralen visade kornigt positivt ANA-fynd med stark positivitet för anti-Jo-1. Ny lungröntgen visade ökad retikulär teckning med misstanke om fibros och interstitiell lungsjukdom. Vid inkomst till reumatologisk avdelning var patienten muskelsvag, men hade inga tecken på artrit eller »mekanikerhänder«. Muskelenzymer var förhöjda. SR var lätt stegrat, men CRP var näst in till normalt.
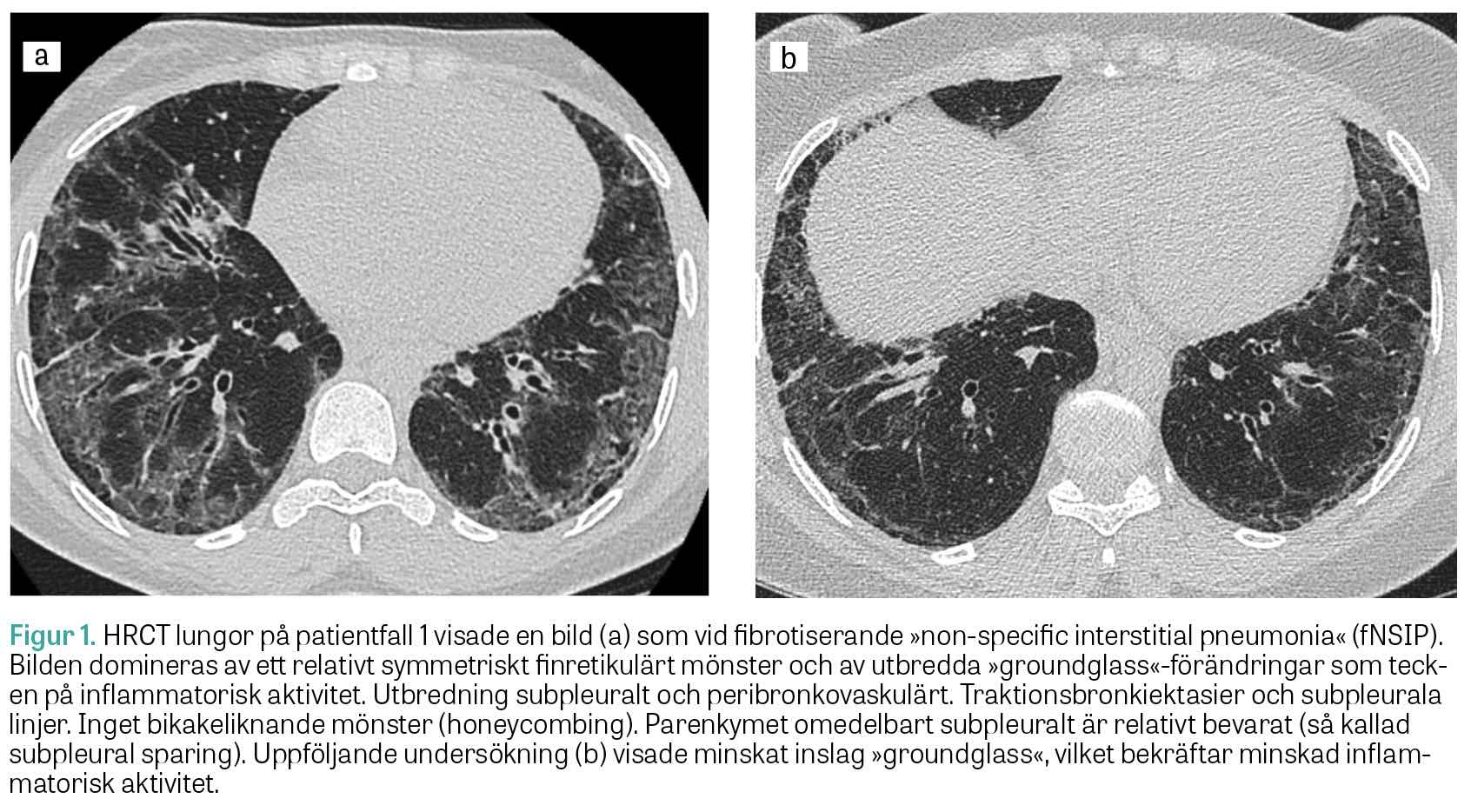
Muskelbiopsi visade histopatologisk bild med perifascikulära patologiska fynd överensstämmande med antisyntetassyndrom. EMG var normalt. MR helkropp visade utbredd inflammation i skelettmuskulatur. HRCT lungor visade mönster av fibrotisk »non-specific interstitial pneumonia« (fNSIP) (Figur 1a). Spirometriundersökningen visade markerad restriktiv lungfunktionsnedsättning: TLC 60 procent, DLCO 49 procent och FVC 52 procent av förväntad funktion.
Två år efter symtomdebut kunde diagnosen anti-Jo-1-positivt antisyntetassyndrom med muskel- och lungengangemang ställas. Patienten behandlades med metylprednisolon (500 mg × 1) i 3 dagar, följt av prednisolon (50 mg × 1) och mykofenolatmofetil (1 g × 2), samt 2 infusioner intravenöst med rituximab (1 000 mg). Vid uppföljning ett år senare hade patienten förbättrats kliniskt, men spirometri visade oförändrad bild med TLC 63 procent, DLCO 53 procent och FVC 60 procent av förväntad funktion. HRCT lungor visade en minskning av både fibrosen och den inflammatoriska aktiviteten (Figur 1b).
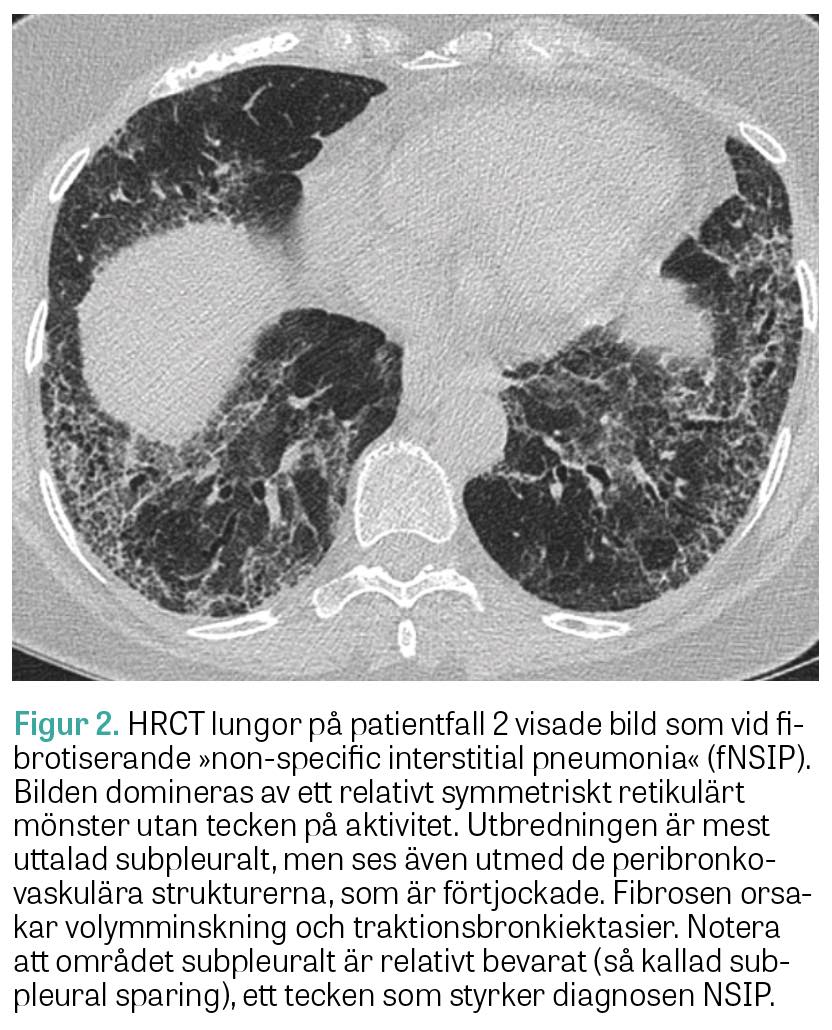
Fall 2. Den andra patienten är en icke-rökande tidigare frisk 65-årig kvinna, som insjuknade med hosta och andfåddhet och remitterades från vårdcentralen till lungmedicinsk klinik för utredning. HRCT lungor visade bild av NSIP. Bronkoskopi och bronkoalveolärt lavage utfördes utan att man såg tecken på infektion. Autoimmun serologi visade negativa ANA och positiva anti-Ro52-antikroppar. Spirometri visade TLC 65 procent, DLCO 45 procent och FVC 67 procent av förväntad funktion. Hon fick påbörja behandling med prednisolon (30 mg/dag), utan nämnvärd klinisk förbättring. Ny HRCT lungor visade påtaglig progress av fibros och även »ground glass«-förändringar förenliga med pågående inflammatorisk aktivitet. Spirometri visade oförändrade värden. Vid diskussion på en multidisciplinär konferens föreslogs komplettering med myositantikroppar, som visade stark positivitet för anti-PL-12 och anti-Ro52. Diagnosen anti-PL-12-positivt antisyntetassyndrom med isolerat lungengagemang kunde fastställas 12 månader efter symtomdebut.
Behandling inleddes med metylprednisolon (500 mg) intravenöst i 3 dagar, följt av prednisolon (50 mg × 1) samt cyklofosfamid (500 mg) intravenöst varannan vecka som på grund av levertoxicitet fick bytas mot infusioner med rituximab (1 000 mg) intravenöst med 2 veckors mellanrum. Underhållsbehandling gavs sedan med mykofenolatmofetil och prednisolon. Efter insatt behandling upplevde patienten minskad andfåddhet och hosta. Uppföljande HRCT lungor var utan inflammatorisk aktivitet eller tecken på progress av fibros. Spirometri två år senare visade förbättrade värden med TLC 74 procent, DLCO 64 procent och FVC 89 procent av förväntad funktion. Prednisolonbehandlingen utsattes då sjukdomsaktiviteten bedömdes som låg.
Fall 3. Den tredje patienten är en icke-rökande man med kronisk njursvikt född på 50-talet. Patienten insjuknade inom loppet av 3 dagar med dyspné, hosta och hemoptys. Han sökte initialt vårdcentral där CRP var kraftigt förhöjt (118 mg/l). Han insattes på doxycyklin med misstanke om luftvägsinfektion. Dagen efter sökte patienten igen på grund av tilltagande besvär med feber och inlades på medicinavdelning. Han förflyttades till Iva med bild av akut svår lungsvikt och intuberades. Datortomografi av lungor visade utbreda bilaterala infiltrat (radiologiska bilder var dock svårtolkade på grund av komorbiditet och visas därför inte i artikeln). Behandling påbörjades med bredspektrumantibiotika utan klinisk förbättring, och odlingar var negativa. Immunserologi visade negativa ANA, men utvidgad serologi inklusive myositpanel påvisade antikropparna anti-Ku och anti-SS-B. Diagnosen myositöverlappningssyndrom med interstitiell pneumonit kunde fastställas.
Behandling påbörjades med infusioner med metylprednisolon (500 mg × 1) i 3 dagar följt av prednisolon (50 mg × 1) och infusion med rituximab (1 000 mg) intravenöst med 2 veckors mellanrum. Patienten förbättrades långsamt och kunde dekanyleras efter 5 veckor. Underhållsbehandling gavs med mykofenolatmofetil (250 mg) dagligen, en låg dos på grund av njursvikt, och infusion med rituximab (1 000 mg) intravenöst var 6:e månad. Prednisolon trappades ner och var helt utsatt 14 månader efter diagnos. Spirometri 2 och 5 månader efter diagnos visade TLC 52 respektive 66 procent, DLCO 45 respektive 58 procent, samt FVC 61 respektive 76 procent. HRCT lungor 12 månader efter diagnos visade endast lätta kvarstående »ground glass«-förändringar.
Sammanfattning
Vi har beskrivit tre typiska fall, där patienter debuterat med lungsymtom som första, och i två fall som enda, symtom. Så småningom kunde diagnosen interstitiell lungsjukdom med myositantikroppar fastställas.
För patient 1 och 2 tog det 2 respektive 1 år från symtomdebut till dess att diagnosen interstitiell lungsjukdom med myosit fastställdes och adekvat behandling påbörjades. I det första fallet fanns misstanke om interstitiell lungsjukdom, men HRCT utfördes inte initialt då patienten avbokade undersökningen. Först två år senare, när muskelsvaghet hade tillkommit, uppstod misstanke om inflammatorisk systemsjukdom och serologisk testning utfördes. Man kunde då fastställa diagnosen antisyntetassyndrom baserat på förekomst av anti-Jo-1. HRCT lungor visade då mönster av NSIP, som är typiska för antisyntetassyndrom. I fall 2 remitterades patienten till lungmedicin, där interstitiell lungsjukdom påvisades vid HRCT-undersökning av lungorna. Eftersom ANA var negativa utfördes testning för myositspecifika antikroppar först ett år senare. Man kunde då påvisa anti-PL-12 och diagnosen anti-PL-12 positivt antisyntetassyndrom kunde fastställas. För patient 3 var förloppet mycket kortare från symtom till diagnos. Trots negativa ANA testade man omgående för autoantikroppar associerade med myosit och fann förekomst av anti-Ku. Patienten fick diagnosen myositöverlappningssyndrom med interstitiell pneumonit och kunde starta immunmodulerande behandling.
Sammanfattningsvis insjuknade dessa patienter med lungmanifestation som första och ibland enda symtom, och ANA var negativ hos två av tre. Serologisk testning av myositantikroppar var helt avgörande för att kunna fastställa diagnos och påbörja adekvat immunmodulerande behandling i samtliga tre fall. Huruvida tillståndet interstitiell lungsjukdom med myositantikroppar och utan muskel- eller hudsymtom ska kallas myosit eller inte debatteras. Sjukdomsbilden har föreslagits få namnet pneumomyosit [29].
Potentiella bindningar eller jävsförhållanden: Ann Mari Svensson har föreläst för Astra Zeneca och Boehringer Ingelheim mot ersättning. Magnus Sköld har medverkat i rådsgrupper, erhållit föreläsningsarvode och/eller forskningsanslag från Astra Zeneca, Boehringer Ingelheim, Chiesi, Pfizer, Roche och Novartis.
(uppdaterad 2022-08-17)
Referenser
- Assayag D, Kim EJ, Elicker BM, et al. Survival in interstitial pneumonia with features of autoimmune disease: a compar-ison of proposed criteria. Respir Med. 2015;109(10):1326-31.
- Oldham JM, Adegunsoye A, Valenzi E, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47(6):1767-75.
- Shao T, Shi X, Yang S, et al. Interstitial lung disease in connective tissue disease: a common lesion with heterogeneous mechanisms and treatment considerations. Front Immunol. 2021;12:684699.
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44-68.
- Bodolay E, Csiki Z, Szekanecz Z, et al. Five-year follow-up of 665 Hungarian patients with undifferentiated connective tissue disease (UCTD). Clin Exp Rheumatol. 2003;21(3):313-20.
- Fischer A, Antoniou KM, Brown KK, et al. ERS/ATS Task Force on Undifferentiated Forms of CTD-ILD. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976-87.
- Svensson J, Arkema EV, Lundberg IE, et al. Incidence and prevalence of idiopathic inflammatory myopathies in Sweden: a nationwide population-based study. Rheumatology (Oxford). 2017;56(5):802-10.
- Meyer A, Meyer N, Schaeffer M, et al. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford). 2015;54(1):50-63.
- Ghirardello A, Doria A. New insights in myositis-specific autoantibodies. Curr Opin Rheumatol. 2018;30(6):614-22.
- McHugh NJ, Tansley SL. Autoantibodies in myositis. Nat Rev Rheumatol. 2018;14(5):290.
- Lundberg IE, De Visser M, Werth VP. Classification of myositis. Nat Rev Rheumatol. 2018;14(5):269.
- Jablonski R, Bhorade S, Strek ME, et al. Recognition and management of myositis-associated rapidly progressive interstitial lung disease. Chest. 2020;158(1):252-63.
- Vij R, Noth I, Strek ME. Autoimmune-featured interstitial lung disease: a distinct entity. Chest. 2011;140(5):1292-9.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity. 2006;39(3):217-21.
- Satoh M, Tanaka S, Ceribelli A, et al. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol. 2017;52(1):1-19.
- Sambataro D, Sambataro G, Pignataro F, et al. Patients with interstitial lung disease secondary to autoimmune diseases: how to recognize them? Diagnostics (Basel). 2020;10(4):208.
- Hallowell RW, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies and the antisynthe-tase syndrome: recent advances. Curr Opin Rheumatol. 2014;26(6):684-9.
- Hervier B, Devilliers H, Stanciu R, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev. 2012;12(2):210-7.
- Trallero-Araguás E, Grau-Junyent JM, Labirua-Iturburu A, et al; IIM Study Group and Autoimmune Diseases Study Group (GEAS) of the Spanish Society of Internal Medicine (SEMI). Clinical manifestations and long-term outcome of anti-Jo1 antisynthe-tase patients in a large cohort of Spanish patients from the GEAS-IIM group. Semin Arthritis Rheum. 2016;46(2):225-31.
- Mecoli CA, Christoph-er-Stine L. Management of interstitial lung disease in patients with myositis specific autoantibodies. Curr Rheumatol Rep. 2018;20(5):27.
- Labirua A, Lundberg IE. Interstitial lung disease and idiopath-ic inflammatory myopathies: progress and pitfalls. Curr Opin Rheumatol. 2010;22(6):633-8.
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60(7):2193-200.
- Matsushita T, Mizumaki K, Kano M, et al. Antimelanoma differentiation-associated protein 5 antibody level is a novel tool for monitoring disease activity in rapidly progressive interstitial lung disease with dermatomyositis. Br J Dermatol. 2017;176(2):395-402.
- Mahler M, Raijmakers R. Novel aspects of autoantibodies to the PM/Scl complex: clinical, genetic and diagnostic insights. Autoimmun Rev. 2007;6(7):432-7.
- Rigolet A, Musset L, Dubourg O, et al. Inflammatory myopathies with anti-Ku antibodies: a prognosis dependent on associated lung disease. Medicine. 2012;91(2):95-102.
- Barsotti S, Lundberg IE. Current treatment for myositis. Curr Treatm Opt Rheumatol. 2018;4(4):299-315.
- Oddis CV, Aggarwal R. Treatment in myositis. Nat Rev Rheumatol. 2018;14(5):279-89.
- Shappley CL, Paik JJ, Saketkoo LA. Myositis-related interstitial lung diseases: diagnostic features, treatment, and complications. Curr Treatm Opt Rheumatol. 2019;5(1):56-83.
- Danoff SK. Myositis: prognostic factors in »pneumo-myositis«. Nat Rev Rheumatol. 2018;14(5):253-4.
Summary
Interstitial lung disease can be the first sign of systemic autoimmune disease. If associated with myositis, interstitial lung disease may be the only symptom, with no presence of muscular weakness or other extramuscular manifestations. ANA-testing performed with indirect immune fluorescence may be negative. Testing for myositis antibodies should be considered as a step in the diagnostic process when strong clinical suspicion of interstitial lung disease of unknown origin is present.
If interstitial lung disease is suspected, the patients should be referred to a specialist clinic for further investigation and, if possible, for discussion within a multidisciplinary team. Early suspicion of systemic inflammatory disease, rapid diagnosis and early start of treatment are crucial for future prognosis, quality of life and survival.