Om Aβ-immunterapi vid Alzheimers sjukdom blir godkänt för kliniskt bruk kommer det att krävas en säker och kostnadseffektiv diagnostik som fungerar både i primärvård och sekundärvård.
Nivån av fosforylerat tau-protein i plasma är tydligt ökad vid alzheimer, men normal vid andra neurodegenerativa sjukdomar, och den variant som kallas P-tau-217 ger högst diagnostisk säkerhet.
En ny hjärnspecifik variant av tau-protein (BD-tau) visar lovande resultat som markör för neurodegeneration av alzheimertyp.
Blodtesten skulle kunna användas i primärvården för att identifiera patienter med tidiga kognitiva symtom som bör remitteras till specialistklinik eller minnesmottagning, och med en tvåstegsmodell för testning uppnås en hög säkerhet i att förutsäga eller utesluta hjärnamyloidos.
Vi har bevittnat imponerande framsteg inom den kliniska forskningen kring Alzheimers sjukdom, vilka gett en detaljerad kunskap om den molekylära patologi som karakteriserar sjukdomen. Dessa patofysiologiska processer innefattar i korthet aggregering av amyloid-beta (Aβ) till Aβ-innehållande plack, fosforylering och aggregering av tau-protein med bildning av neurofibriller i nervcellernas cytoplasma och dystrofiska neuriter som omger placken, samt degeneration av nervceller och deras synapser [1]. Denna kunskap har omsatts till riktade biokemiska test i likvor (ryggmärgsvätska), där man finner en minskad Aβ42-koncentration tillsammans med sänkt Aβ42/40-kvot (indikerande Aβ-innehållande plack), ökning av fosforylerat tau-protein (P-tau), som indikerar tau-patologi, samt en ökning även av »totalt« tau-protein (T-tau) som tecken på neurodegeneration och synapsmarkörer som neurogranin, vilka indikerar synapsdysfunktion eller -degeneration [2, 3]. Dessa alzheimertest har genomgått en mycket omfattande analytisk och klinisk validering och finns i dag tillgängliga för klinisk rutindiagnostik som FDA-godkända test med helautomatiserade instrument som ger en mycket hög diagnostisk precision [4, 5], vilket är verifierat i en lång rad studier [3, 6]. Utöver likvormarkörer används i dag positronemissionstomografi (PET) för att påvisa förekomst av Aβ-innehållande plack (Aβ-PET) och tau-innehållande neurofibriller (tau-PET) i hjärnan.
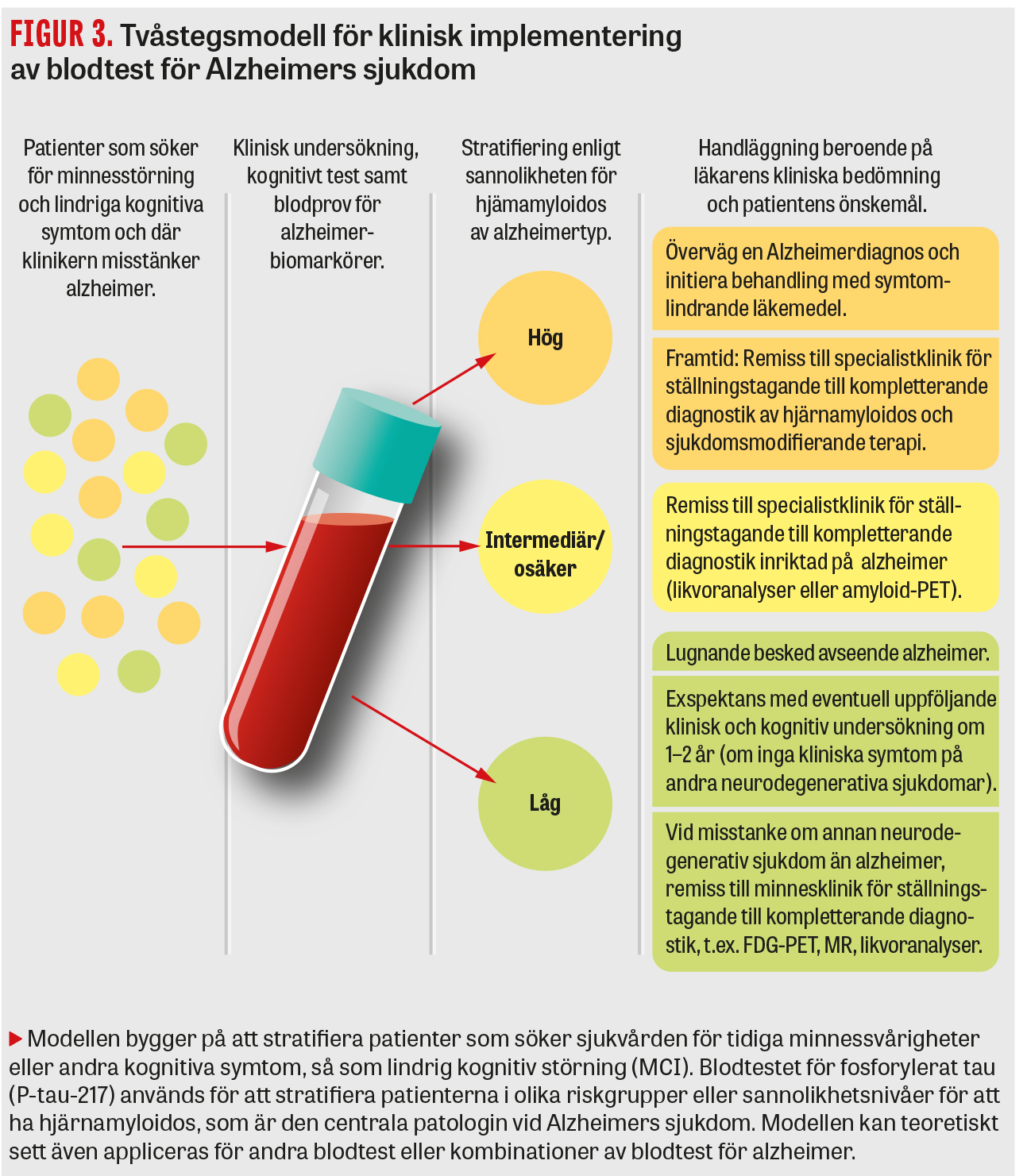
Ett annat framsteg inom alzheimerforskningen är utvecklingen av immunterapi riktad mot Aβ. De monoklonala antikropparna lecanemab [7] och donanemab [8] har i stora fas 3-studier gett en snabb och kraftig minskning av Aβ-innehållande plack i hjärnan tillsammans med en reducerad klinisk progresshastighet, där den kognitiva och funktionella försämringen minskat med 25–40 procent. Dessa läkemedel är i dag under utvärdering för godkännande hos den europeiska läkemedelsmyndigheten EMA för behandling av personer med lindrig kognitiv störning (mild cognitive impairment) eller lindrig demens (ofta definierat som 21–26 poäng på MMSE, Mini mental state examination) på basen av Alzheimers sjukdom. För att säkerställa diagnosen Alzheimers sjukdom kommer det sannolikt att krävas en verifiering med träffsäkra markörer för åtminstone Aβ-patologi [9]. Inför introduktionen av dessa läkemedel vore mer lättillgängliga och billigare biomarkörer önskvärda, eftersom PET-undersökning endast utförs vid högspecialiserade centrum och innefattar en exponering för strålning, medan rutinmässig lumbalpunktion kräver träning och rutin och därmed kan vara svår att introducera på vårdcentraler i större skala. De blodtest för alzheimer som utvecklats bara de senaste åren kommer därför sannolikt att spela en viktig roll i vårdkedjan och den kliniska handläggningen av patienter med tidiga minnessvårigheter och andra kognitiva symtom där man misstänker Alzheimers sjukdom [10]. Vi presenterar en tvåstegsmodell där blodbiomarkörer används tillsammans med klinisk undersökning och en enkel kognitiv testning i ett första steg vid utredning av patienter med kognitiva symtom [11]. Informationen om risken att patienten har hjärnamyloidos (det vill säga alzheimerpatologi) används för vägledning om vilka patienter som bör remitteras till specialistkliniken för en mer detaljerad klinisk utredning, där likvortest eller PET-skanning i steg två kan användas för att verifiera diagnosen Alzheimers sjukdom inför ställningstagande till terapi [11].
Blodtest för Alzheimers sjukdom
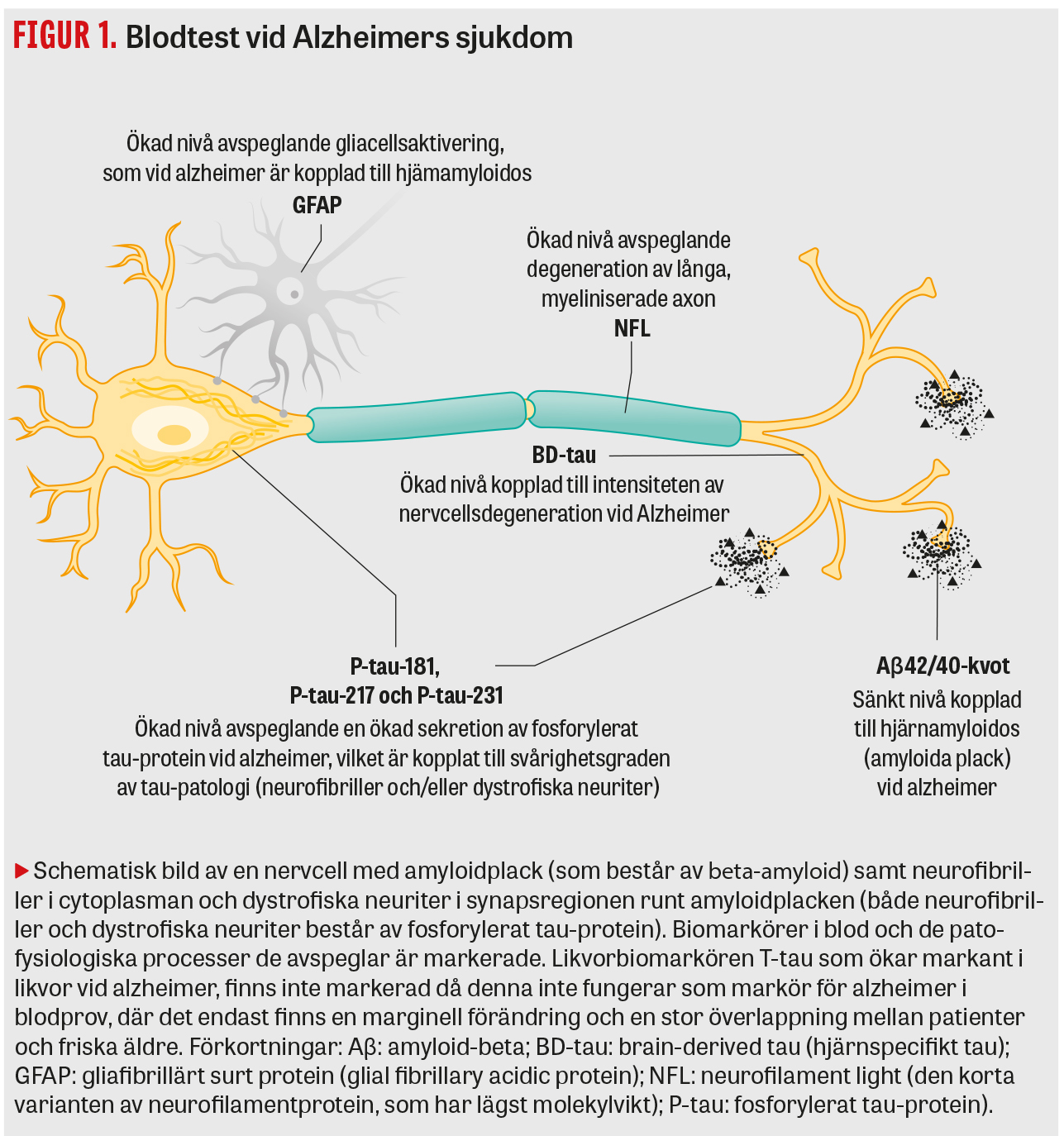
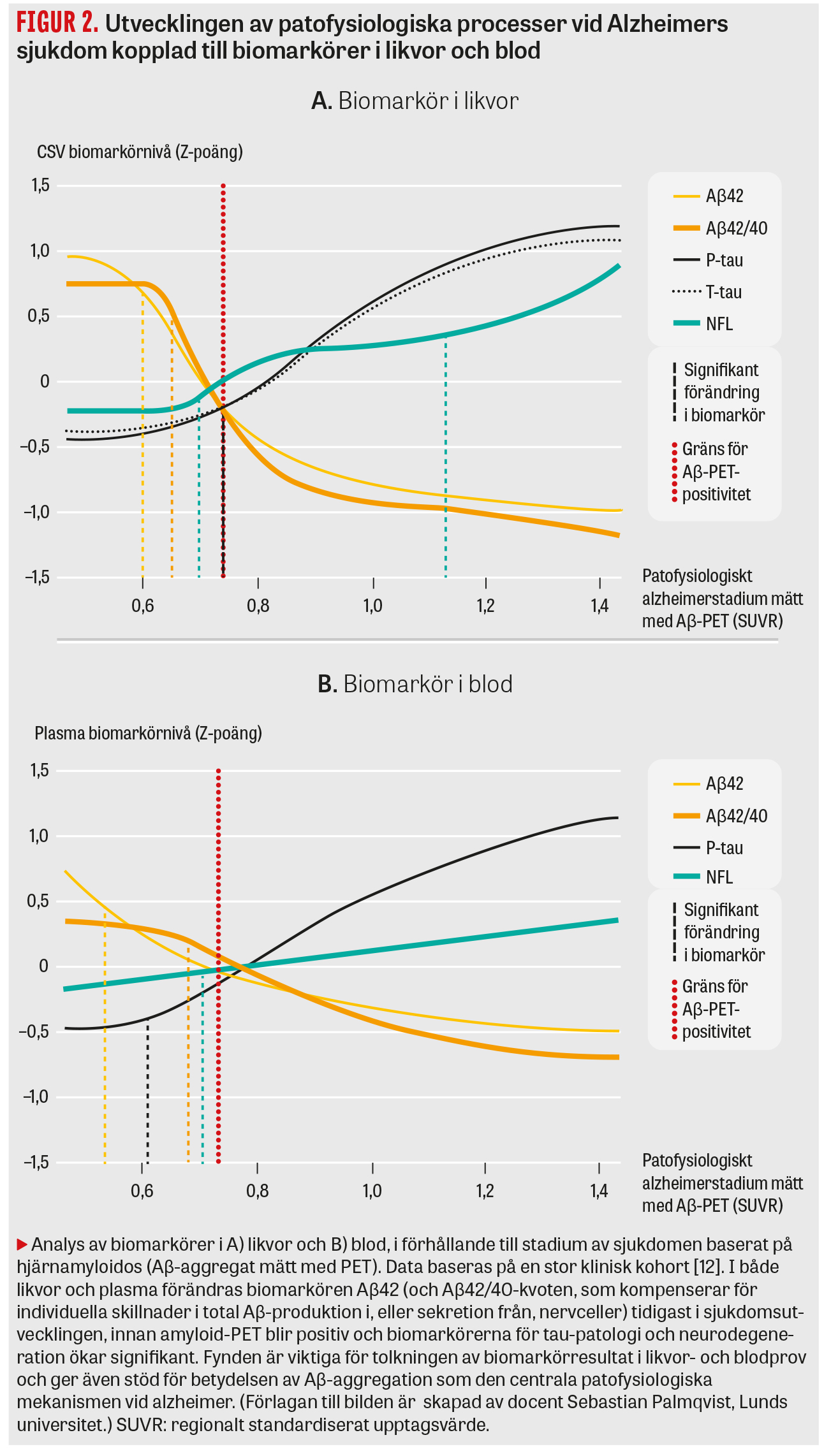
I denna översikt ger vi den vetenskapliga bakgrunden till de blodmarkörer för Alzheimer som utvecklats de senaste åren och som utvärderats i stora kliniska studier. Blodtesten avser i princip samma biomarkörer som utvecklats för likvortestning, det vill säga Aβ42/40-kvot för hjärnamyloidos, P-tau för neurofibriller och tau-patologi, samt T-tau, hjärnspecifikt tau (brain-derived tau, BD-tau) och neurofilament light (NFL) för neurodegeneration (Figur 1). En samstämmig litteratur visar att det är biomarkörerna för hjärnamyloidos som förändras tidigast i sjukdomsförloppet (Figur 2), före biomarkörerna som avspeglar tau-patologi och neurodegeneration [12], vilket är en viktig kunskap vid tolkningen av biomarkörresultat i både likvor- och blodprov. Vi presenterar även en tvåstegsmodell för applicering och tolkning av blodmarkörer vid den initiala handläggningen av patienter med tidiga kognitiva symtom på vårdcentraler eller andra sjukvårdsenheter utanför högspecialiserade universitetskliniker (Figur 3).
Amyloid-beta i plasma
Den 42 aminosyror långa varianten av amyloid-beta (Aβ42) är huvudkomponenten i plack vid Alzheimers sjukdom, då den är mer hydrofob och har en större tendens att aggregera än andra Aβ-former, som Aβ40, vilken produceras i störst mängd. Vid alzheimer aggregerar Aβ42 och bildar plack i hjärnan, vilket leder till att nivån i likvor blir onormalt låg [6], och via en kvot med Aβ40 (Aβ42/40-kvoten) som kompenserar för individuella skillnader i den »totala« Aβ-produktionen blir träffsäkerheten mycket hög för att påvisa hjärnamyloidos [3, 4].
Utvecklingen av den ultrakänsliga immunanalystekniken Simoa [13] och immunprecipitation kombinerat med masspektrometri (IP-MS) [14], metoder för att mäta Aβ42-nivån i plasma, gav hopp om en blodmarkör för hjärnamyloidos. I en första stor klinisk studie kunde man visa att Aβ42/40-kvoten är sänkt i plasma vid alzheimer, även i den lindriga fasen av sjukdomen som kallas lindrig kognitiv störning (mild cognitive impairment, MCI), och att nivån var kopplad till hjärnamyloidos mätt med Aβ-PET [15]. Dessa fynd har verifierats med annan metodik, inkluderande helautomatiserade högprecisionsinstrument [16] och IP-MS [17, 18], med mycket liknande resultat.
Trots lovande resultat på gruppnivå finns det problem med att använda plasma-Aβ för att påvisa hjärnamyloidos hos enskilda patienter på det sätt som görs i klinisk rutin. Anledningen är att sänkningen av plasma-Aβ42/40-kvoten hos Aβ-PET-positiva individer är betydligt mindre i plasma (10–15 procent) än i likvor (cirka 50 procent) [19, 20], vilket leder till en närmast fullständig överlappning mellan alzheimerpatienter och friska, och sannolikt beror på att en betydande del av Aβ i plasma kommer från perifera vävnader och endast en mindre andel från hjärnan. För detta talar till exempel fyndet från många studier att korrelationen mellan plasma- och likvornivåer av Aβ är mycket låg eller obefintlig [20, 21]. Flera studier har visat att den stora överlappningen mellan Aβ-PET-positiva och -negativa patienter leder till att plasma-Aβ42/40-kvoten har en mycket låg klinisk robusthet, det vill säga en alltför låg förmåga att tolerera den provrelaterade eller analytiska variabilitet som alltid ses för biomarkörer som används regelbundet i klinisk rutin, vilket skulle leda till feldiagnostik av patienter [22, 23]. Detta har medfört att stora internationella diagnostikaföretag valt att inte utveckla denna biomarkör vidare för klinisk användning. Plasma-Aβ42/40-kvoten kan givetvis ändå vara värdefull i klinisk forskning och i läkemedelsprövningar, där jämförelser görs på gruppnivå.
Fosforylerat tau-protein i plasma
Vid Alzheimers sjukdom hyperfosforyleras tau-protein och aggregerar till dels neurofibriller (tangles) inne i de påverkade nervcellernas cytoplasma, dels i trådliknande strukturer i axoner och dendriter (neuropil threads) liksom i uppdrivna nervändslut (dystrophic neurites), där de senare omringar de Aβ-innehållande placken och sannolikt uppkommer som en reaktion på Aβ-aggregaten [24]. Förutom vid Alzheimers sjukdom ses olika former av tau-patologi vid andra neurodegenerativa sjukdomar som kollektivt går under benämningen »tauopatier« och inkluderar till exempel progressiv supranukleär paralys (PSP), kortikobasal degeneration (CBD) och varianter av frontotemporal demens (FTD) [25], men mängden tau-aggregat är generellt sett mycket lägre vid dessa sjukdomar än vid alzheimer.
Det finns ett mycket stort antal studier som visar att P-tau-koncentrationen i likvor är ökad vid Alzheimers sjukdom. Denna ökning är specifik för alzheimer; andra demenssjukdomar och även tauopatier har normala nivåer [2, 26], och graden av ökning av P-tau i likvor är associerad med svårighetsgraden av tau-patologi mätt med PET, men predicerar även kommande spridningsgrad av tau-patologin [27-29].
I dag kan man även mäta P-tau i plasmaprov, och bara de senaste åren har en lång rad studier samstämmigt visat att det finns en klar ökning av flera olika varianter av P-tau vid Alzheimers, sjukdom inkluderande P-tau-181, P-tau-217 och P-tau-231 [10, 30-33]. Ökningen av P-tau i plasma ses även i den lindriga MCI-fasen av Alzheimers sjukdom, men däremot är nivån normal vid andra tauopatier, såsom FTD, PSP och CBD [33, 34]. I studier där man jämfört dessa olika former av P-tau har man funnit att både P-tau-181 och P-tau-217 har en mycket hög samstämmighet med Aβ-PET och tau-PET [35] och att P-tau-181, P-tau-217 och P-tau-231 korrelerar tätt och har en likartad hög förmåga att identifiera alzheimer [36]. En viktig skillnad, återkommande i olika studier, är att graden av ökning (skillnaden i medelvärde mellan grupper av alzheimerpatienter och kontroller) är klart högre för P-tau-217 än för de andra varianterna vid symtomatisk alzheimer, vilket ger en större robusthet och diagnostisk säkerhet då analysen introduceras i klinisk diagnostik [10]. Utöver att plasma-P-tau med hög säkerhet kan skilja alzheimer från andra hjärnsjukdomar i patientpopulationer med demens, finns det studier som pekar mot att plasma-P-tau-217 på gruppnivå kan predicera vilka patienter med lindriga minnesbesvär som kommer utveckla demens på basen av alzheimer inom de kommande 2–6 åren [37, 38]. Denna prediktion blir ännu bättre om plasma-P-tau-217 kombineras med några korta kognitiva test, som kan genomföras på 15–20 minuter även inom primärvården [38].
Ytterligare studier där man jämfört olika former av P-tau i plasma hos symtomfria äldre och via longitudinella prov har funnit att plasma-P-tau-231 ökar tidigast i förloppet, med lägre grad av hjärnamyloidos, medan P-tau-217 ökar mest över en period av 4–6 år, både i den prekliniska och den symtomatiska fasen av sjukdomen [30, 39, 40]. Vidare kan plasma-P-tau-217 även predicera vilka symtomfria äldre personer med preklinisk alzheimer som kommer att ansamla tau-innehållande neurofibriller samt vilka som kommer att försämras kognitivt inom de närmaste 4–6 åren [28, 41]. Sammantaget är plasma-P-tau, framför allt P-tau-217, en mycket lovande alzheimermarkör, och vissa studier pekar på att detta test på gruppnivå kan kan påvisa alzheimerpatologi i hjärnan med nästan lika stor träffsäkerhet som kliniskt använda likvor- och PET-baserade test [10].
Markörer för neurodegeneration
Genom att mäta nivån av nervcellsproteiner i likvor eller plasma kan man få en indikation om intensiteten av nervcellsdegeneration eller svårighetsgraden av en akut nervcellsskada. Denna information kan fungera som ett komplement till DT och MR och ge ett mått på graden av atrofi, det vill säga till vilket stadium nervcellsdegenerationen kommit.
Neurofilament light. Neurofilament light (NFL) är ett av de intermediärfilament som bygger upp nervcellernas cytoskelett och finns huvudsakligen i grova myeliniserade axon i CNS. NFL utsöndras till likvor och blod under fysiologiska förhållanden, där det fungerar som en känslig, men icke-sjukdomsspecifik biomarkör för axonal nervcellsskada; ökade nivåer ses både vid olika typer av akuta hjärnskador (till exempel vid hjärntrauma) och vid flera olika neurodegenerativa sjukdomar [42, 43]. Introduktionen av NFL i plasma mätt med Simoa-teknik blev ett viktigt steg mot en ökad tillgänglighet, inte minst inom primärvården, då redan den första studien visade att nivån av NFL i plasma korrelerar mycket väl med nivån i likvor [44], vilket talar för att nivån i plasma avspeglar patofysiologiska processer i CNS.
Studier har visat att plasma-NFL är klart ökat vid Alzheimers sjukdom, och högt plasma-NFL kan predicera snabbare kognitiv försämring och progresshastighet av kortikal atrofi mätt med MR-teknik [45]. Vidare visar longitudinella data att plasma-NFL ökar snabbare med mer uttalad neurodegeneration och mer uttalad kognitiv försämring, talande för att det inte bara avspeglar intensiteten utan även graden av neurodegeneration [46]. Ett viktigt fynd är också att ökningen av plasma-NFL kan ses redan i den prekliniska fasen av sjukdomen, mellan 5 och 20 år före de första symtomen, vilket visats i ett flertal studier på familjär alzheimer [47, 48].
Ökad NFL-koncentration i plasma är dock inte specifik för alzheimer utan ses även vid flera andra neurodegenerativa sjukdomar, där ökningen ofta även är mer uttalad [43]. Mycket kraftigt ökat plasma-NFL ses exempelvis vid Creutzfeldt–Jakobs sjukdom [49], där det föreligger en mycket intensiv neurodegeneration och därmed klinisk symtomprogress. Ett annat kliniskt användningsområde är frontotemporal demens, där plasma-NFL-nivån är nästan fyra gånger högre än hos kontroller, framför allt i gruppen med primär progressiv afasi [50]. Plasma-NFL är också kliniskt användbart som stöd i differentiering mellan klassisk Parkinsons sjukdom, där nivån är normal, och andra parkinsonistiska sjukdomar som PSP, MSA och CBD, som uppvisar tydligt ökade värden [43, 51]. En annan indikation är amyotrofisk lateral skleros (ALS), där NFL-nivån är kraftigt ökad och har en mycket hög diagnostisk säkerhet [52, 53], och kan predicera klinisk progress och överlevnad vid sjukdomen [54].
Plasma-NFL ökar även vid akuta hjärnskador såsom akut traumatisk hjärnskada (traumatic brain injury, TBI), där skada på nervcellernas långa axon är en viktig mekanism [55]. Efter måttlig–svår TBI ses en uttalad ökning av NFL i blod [56]. Ökningen av NFL i plasma är relativt långsam och utdragen, med en mindre ökning de första dagarna efter skadan (förutom i allvarliga fall, då ökningen kan ses redan efter ett dygn), och högst nivåer 5–12 dagar efter skadan [56]. Graden av ökning av NFL i blod predicerar klinisk långtidsprognos [56]. Ökat NFL i blod ses även vid lindrig TBI (hjärnskakning), där man exempelvis hos amatörboxare funnit en klar ökning av NFL efter match, som kan kopplas samman med antalet träffar mot huvudet och som normaliseras i lugnt skede [57]. NFL ökar även vid hypoxisk hjärnskada efter hjärtstopp, och stora prospektiva studier har visat att ökat NFL i blod har ett mycket högt prediktionsvärde för dåligt kliniskt utfall även då prov tas så tidigt som första dagen efter hjärtstoppet. NFL i blod fungerade klart bättre än andra blodmarkörer (NSE och S100B), och markant bättre än rutinmässiga kliniska undersökningar som EEG, datortomografi av hjärnan eller kliniska test som pupill- och kornealreflexer [58].
Plasma-tau-protein
Med de nya analysteknikerna kan även T-tau mätas i blodprov. Den första Simoa-metoden för T-tau presenterades 2013 och visade en nästan 1 000-faldigt bättre analytisk känslighet än de ELISA-metoder (enzymkopplad immunadsorberande analys) som användes för att mäta T-tau i likvor [59]. Studier av den diagnostiska potentialen hos plasma-T-tau har dock gett nedslående resultat, med endast ett marginellt ökat medelvärde på gruppnivå, och i stället en markant överlappning mellan alzheimerpatienter och kognitivt normala äldre [60]. Förklaringen till att T-tau fungerar väl som alzheimermarkör i likvor, men inte i blod, är sannolikt att tau-protein även bildas i perifera nerver och vävnader och utsöndras till plasma, men de analysmetoder som använts kan inte skilja mellan tau som bildats i hjärnan och i perifera vävnader.
Av detta skäl utvecklades en ny Simoa-metod för hjärnspecifikt tau (brain-derived tau, BD-tau), en antikropp som endast reagerar med tau som bildats i hjärnan [61, 62]. I motsats till tau visar denna metod en stark korrelation mellan nivåer i parade blod- och likvorprov. Vid alzheimer har man i flera olika kohorter funnit en mycket tydlig ökning av plasma-BD-tau, även i tidiga faser av sjukdomen, medan nivån var normal vid andra neurodegenerativa sjukdomar, och i studier med neuropatologisk uppföljning ses ett tydligt samband med alzheimerpatologi, men inte andra former av patologi [62]. Ökningen av plasma-BD-tau är ännu mer uttalad vid Creutzfeldt–Jakobs sjukdom och andra snabbt progredierande demenssjukdomar än vid alzheimer [61], talande för att BD-tau i blod kan fungera som en värdefull biomarkör för intensiteten av neurodegeneration [63].
Markörer för gliacellsaktivering
Vid en rad hjärnsjukdomar, inklusive alzheimer, ses en astrocytaktivering och astrocytos, och i samband med detta bildar astrocyterna en ökad mängd gliafibrillärt surt protein (glial fibrillary acidic protein, GFAP). Likvor-GFAP har sedan många år funnits tillgänglig som biomarkör för astrocytos eller astrocytaktivering. Ökade nivåer ses till exempel vid multipel skleros, där ökningen korrelerar med neurologiskt deficit [64], och vid infektioner i CNS såsom borreliaencefalit, där nivån även minskar efter adekvat antibiotikabehandling [65], men även vid Alzheimers sjukdom är nivån klart ökad jämfört med åldersmatchade kontroller [66].
Med ELISA-metodik har man också visat att GFAP i blod ökar vid akuta hjärnåkommor som subaraknoidalblödning [67] och akut traumatisk hjärnskada [68], och graden av ökning predicerar både kliniskt och neuroradiologiskt status vid 1-årsuppföljning. GFAP i plasma mätt med den nya Simoa-tekniken har visats vara av intresse även vid kroniska neurodegenerativa sjukdomar där ökningen är mindre uttalad än vid akuta svåra hjärnskador. Vid symtomatisk alzheimer (lindrig kognitiv störning eller demens) är nivån klart högre än hos Aβ-negativa äldre [69, 70], och en lätt ökning av plasma-GFAP ses redan hos kognitivt normala äldre som har hjärnamyloidos påvisad med Aβ-PET [69, 71].
Då man funnit en ökning av plasma-GFAP hos kognitivt normala äldre som har tecken på hjärnamyloidos (mycket tidig alzheimerpatologi) mätt med Aβ-PET, men inte påvisbar tau-patologi mätt med tau-PET [69, 70], har man spekulerat om huruvida plasma-GFAP är en markör för hjärnamyloidos, som utsöndras till blod på grund av att amyloidaggregat aktiverar astrocyterna. Denna hypotes kan vara svår att leda i bevis, eftersom PET-metodiken för att påvisa hjärnamyloidos är känsligare och blir positiv tidigare i sjukdomsförloppet än metoden för tau-PET. Dessutom visar studier att GFAP-nivån i blod är ökad och kopplad till sjukdomsaktiviteten vid andra sjukdomar, som FTD [72] och fästingburen encefalit [73], vilket talar emot att GFAP skulle vara en ren amyloidmarkör.
Ny tvåstegsmodell ökar träffsäkerheten
Som nämnts ovan är det svårt att kraftigt skala upp användningen av PET-baserade alzheimertest för att förbättra diagnostiken, vilket nu börjar bli ett akut problem i många länder. I USA, där de nya immunterapierna mot amyloidos nu implementeras i klinisk praxis, utför dock många läkare utanför universitetssjukhusen av tradition inte lumbalpunktion, vilket som nämnts ovan är alternativet till PET. En korrekt diagnos av Alzheimers sjukdom är viktig inte bara i detta nya sammanhang, utan även för att kunna ge de drabbade och deras anhöriga korrekt information om sjukdomen och dess prognos så att de kan planera sina liv och få tillgång till korrekt symtomlindrande behandling och omsorg. De nya blodtesten, med P-tau-217 i spetsen, kan bli en lösning på detta dilemma, men tidigt i förloppet av alzheimer är risken för falskt positiva resultat relativt stor. Då dessa blodtest inom kort blir tillgängliga för klinisk rutin i Sverige är den mest lämpliga kliniska applikationen som en del i en första bedömning av risken för hjärnamyloidos. Blodtesten skulle då appliceras för patienter som söker sjukvården för minnessvårigheter och andra tidiga kognitiva symtom som skulle kunna vara tidiga tecken på Alzheimers sjukdom. Som visas i Figur 3 är tanken med tvåstegsmodellen att alla patienter med misstänkt alzheimer först genomgår ett P-tau-217-baserat blodtest där sannolikheten för hjärnamyloidos (indikerande alzheimerpatologi) bestäms på individnivå.
I detta första steg av utredningen kan tre utfall nås:
- Mycket hög sannolikhet för alzheimer.
- Mycket låg sannolikhet för alzheimer.
- Intermediära blodvärden, och därmed osäker sannolikhet för alzheimer.
De patienter som har en mycket hög respektive låg sannolikhet för alzheimer behöver ingen vidare utredning för att påvisa eller utesluta eventuell alzheimerpatologi. Patienter med positivt testresultat som kan vara aktuella för immunterapi, liksom patienter med intermediära/osäkra värden, kan remitteras till minnesklinik där de i många fall behöver genomgå likvor- och PET-baserade test i ett andra steg.
Vi har testat denna tvåstegsmodell på två oberoende svenska och en kanadensisk kohort baserade vid minneskliniker, där patienterna hade lindrig kognitiv störning och där läkaren bedömde det viktigt att undersöka och diagnostisera eventuell förekomst av alzheimer [11]. Då vi stipulerade att blodtestet (plasma-P-tau-217) skulle ha 95 procents specificitet och sensitivitet för alzheimerpatologi hos patienter vars sannolikhet för alzheimer klassificerats som mycket hög eller mycket låg, fick 29 procent intermediära/osäkra utfall (då likvortest eller PET krävs för en säker diagnos i ett andra steg) [11]. Med andra ord kunde detta blodtest minska användningen av likvor- och PET-baserade undersökningar med >70 procent, samtidigt som hela tvåstegsmodellen gav 90 procent träffsäkerhet för att detektera alzheimer. Dessutom hade >95 procent av patienterna med hög sannolikhet för alzheimer enligt blodtestet faktiskt alzheimerpatologi i hjärnan [11]. Detta är viktigt eftersom det utöver psykologiska konsekvenser av en felaktig diagnos, till exempel baserad på ett falskt positivt resultat från ett blodtest, också finns andra aspekter på ett test som appliceras på stora patientgrupper för att ställa diagnos och initiera behandling. Det gäller både kostnader och, inte minst, potentiella medicinska risker med behandling för en sjukdom som patienten inte har.
Resultaten från blodtestet visar på en mycket hög säkerhet i identifieringen av både lågriskpatienter, där alzheimerpatologi kan uteslutas, och högriskpatienter, som därmed kan få en diagnos och ges möjlighet att starta en behandling som lindrar symtom. I en framtid skulle högriskpatienter också kunna remitteras till en specialistklinik för eventuell sjukdomsmodifierande behandling. Ju bättre blodtesten blir framöver, desto färre individer kommer att få intermediära/osäkra utfall på dessa test. Minskningen av gruppen med intermediär risk leder i sin tur till ett kraftigt minskat behov av mer avancerad diagnostik med likvortest och PET, vilket skulle förkorta handläggningstiderna och ge stora kostnadsbesparingar inom vården. Denna tvåstegsmodell kan därför vara en kliniskt användbar strategi för implementering av P-tau-217-blodtest. Initialt kan detta göras inom sekundär vård som specialiserade minnesmottagningar, men på sikt även inom primär vård [74]. Just nu pågår studier inom primärvården i Skåne för att studera hur plasma-P-tau-217 kan underlätta diagnostik och handläggning av patienter med kognitiva besvär.
Sammanfattande kommentar
Sammantaget finns det således i dag flera biomarkörer i blod som är kopplade till sjukdomens patofysiologi och som visar en hög förmåga att påvisa Alzheimers sjukdom. Det har i dag utvecklats metoder för att med hög precision mäta biomarkörerna i blodprov med helautomatiska instrument. Fördelen med detta är, förutom enkelheten med analyser på laboratoriet, att denna typ av metoder har en mycket hög precision och att instrumenten finns även på mindre sjukhuslaboratorier. Exempelvis finns det i dag på helautomatiserade högprecisionsinstrument forskningsmetoder för mätning av plasma-Aβ42/40-kvot, P-tau (både P-tau-181 och P-tau-217), ApoE4-nivå för ApoE-fenotypning samt NFL och GFAP. För framtiden, inte minst den dag sjukdomsmodifierande läkemedel finns för behandling av alzheimer i klinisk praxis, kan blodbiomarkörer tillsammans med klinisk undersökning och en enkel kognitiv testning användas för en första screening av patienter med kognitiva symtom och vägledning för vilka patienter som kan remitteras till specialistkliniken för en mer detaljerad klinisk utredning samt likvor- eller PET-biomarkörtest inför ställningstagande till terapi.
Potentiella bindningar eller jävsförhållanden: Kaj Blennow har varit konsult åt Abcam, Axon, Biogen, Lilly, MagQu, Novartis och Roche Diagnostics samt är en av grundarna av Brain Biomarker Solutions in Gothenburg AB, ett GU Ventures-bolag vid Göteborgs universitet. Oskar Hansson har varit konsult åt AC Immune, Amylyx, Alzpath, Bioarctic, Biogen, Cerveau, Eisai, Eli Lilly, Fujirebio, Merck, Novartis, Novo Nordisk, Roche, Sanofi och Siemens.
(uppdaterad 2024-06-03)
Referenser
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387-403.
- Blennow K, Hampel H, Weiner M, et al. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131-44.
- Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27(6):954-63.
- Gobom J, Benedet AL, Mattsson-Carlgren N, et al. Antibody-free measurement of cerebrospinal fluid tau phosphorylation across the Alzheimer’s disease continuum. Mol Neurodegener. 2022;17(1):81.
- Hansson O, Seibyl J, Stomrud E, et al; Swedish BioFINDER study group; Alzhei-mer’s Disease Neuro-imaging Initiative. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14(11):1470-81.
- Blennow K, Mattsson N, Schöll M, et al. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36(5):297-309.
- van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9-21.
- Sims JR, Zimmer JA, Evans CD, et al; TRAILBLAZER-ALZ 2 Investigators. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330(6):512-27.
- Cummings J, Apostolova L, Rabinovici GD, et al. Lecanemab: appropriate use recommendations. J Prev Alzheimers Dis. 2023;10(3):362-77.
- Hansson O, Blennow K, Zetterberg H, et al. Blood biomarkers for Alzheimer’s disease in clinical practice and trials. Nat Aging. 2023;3(5):506-19.
- Brum WS, Cullen NC, Janelidze S, et al. A two-step workflow based on plasma p-tau217 to screen for amyloid β positivity with further confirmatory testing only in uncertain cases. Nat Aging. 2023;3(9):1079-90.
- Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol Med. 2019;11(12):e11170.
- Zetterberg H, Mörtberg E, Song L, et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS One. 2011;6(12):e28263.
- Pannee J, Törnqvist U, Westerlund A, et al. The amyloid-β degradation pattern in plasma – a possible tool for clinical trials in Alzheimer’s disease. Neurosci Lett. 2014;573:7-12.
- Janelidze S, Stomrud E, Palmqvist S, et al. Plasma beta-amyloid in Alzheimer’s disease and vascular disease. Sci Rep. 2016;6:26801.
- Palmqvist S, Janelidze S, Stomrud E, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related beta-amyloid status. JAMA Neurol. 2019;76(9):1060-9.
- Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature. 2018;554(7691):249-54.
- Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13(8):841-9.
- Schindler SE, Bollinger JG, Ovod V, et al. Highprecision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93(17):e1647-59.
- Janelidze S, Teunissen CE, Zetterberg H, et al. Head-to-head comparison of 8 plasma amyloid-beta 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78(11):1375-82.
- Hansson O, Zetterberg H, Vanmechelen E, et al. Evaluation of plasma Abeta(40) and Abeta(42) as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiol Aging. 2010;31(3):357-67.
- Benedet AL, Brum WS, Hansson O, et al; Alzheimer’s Disease Neuro-imaging Initiative. The accuracy and robustness of plasma biomarker models for amyloid PET positivity. Alzheimers Res Ther. 2022;14(1):26.
- Rabe C, Bittner T, Jethwa A, et al. Clinical performance and robustness evaluation of plasma amyloid-beta42/40 pre-screening. Alzheimers Dement. 2023;19(4):1393-402.
- Masters CL, Bateman R, Blennow K, et al. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056.
- Holtzman DM, Carrillo MC, Hendrix JA, et al. Tau: from research to clinical development. Alzheimers Dement. 2016;12(10):1033-9.
- Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med. 2018;284(6):643-63.
- Mattsson-Carlgren N, Andersson E, Janelidze S, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive tau PET in Alzheimer’s disease. Sci Adv. 2020;6(16):eaaz2387.
- Leuzy A, Smith R, Cullen NC, et al. Biomarker-based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2022;79(2):149-58.
- Leuzy A, Janelidze S, Mattsson-Carlgren N, et al. Comparing the clinical utility and diagnostic performance of CSF P-tau181, P-tau217, and P-tau231 assays. Neurology. 2021;97(17):e1681-94.
- Ashton NJ, Pascoal TA, Karikari TK, et al. Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 2021;141(5):709-24.
- Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathol-ogy and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26(3):379-86.
- Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19(5):422-33.
- Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772-81.
- Thijssen EH, La Joie R, Wolf A, et al; Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) investigators. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26(3):387-97.
- Thijssen EH, La Joie R, Strom A, et al; Advancing Research and Treatment for Frontotemporal Lobar Degeneration investigators. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021;20(9):739-52.
- Bayoumy S, Verberk IMW, den Dulk B, et al. Clinical and analytical comparison of six Simoa assays for plasma P-tau isoforms P-tau181, P-tau217, and P-tau231. Alzheimers Res Ther. 2021;13(1):198.
- Cullen N, Leuzy A, Palmqvist S, et al. Individualized prognosis of cognitive decline and dementia in mild cognitive impairment based on plasma biomarker combinations. Nat Aging. 2021;1(1):114-23.
- Palmqvist S, Tideman P, Cullen N, et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat Med. 2021;27(6):1034-42.
- Milà-Alomà M, Ashton NJ, Shekari M, et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat Med. 2022;28(9):1797-801.
- Ashton NJ, Janelidze S, Mattsson-Carlgren N, et al. Differential roles of Aβ42/40, p-tau231 and p-tau217 for Alzheimer’s trial selection and disease monitoring. Nat Med. 2022;28(12):2555-62.
- Mattsson-Carlgren N, Salvadó G, Ashton NJ, et al. Prediction of longitudinal cognitive decline in preclinical Alzheimer disease using plasma biomarkers. JAMA Neurol. 2023;80(4):360-9.
- Lewczuk P, Riederer P, O’Bryant SE, et al; WFSBP Task Force. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: an update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J Biol Psychiatry. 2018;19(4):244-328.
- Ashton NJ, Janelidze S, Al Khleifat A, et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun. 2021;12(1):3400.
- Gisslén M, Price RW, Andreasson U, et al. Plasma concentration of the neurofilament light protein (NFL) is a biomarker of CNS injury in HIV infection: a cross-sectional study. EBioMedicine. 2016;3:135-40.
- Mattsson N, Andreasson U, Zetterberg H, et al; Alzheimer’s Disease Neuroimaging Initia-tive.- Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74(5):557-66.
- Mattsson N, Cullen NC, Andreasson U, et al. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76(7):791-9.
- Quiroz YT, Zetterberg H, Reiman EM, et al. Plasma neurofilament light chain in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional and longitudinal cohort study. Lancet Neurol. 2020;19(6):513-21.
- Weston PSJ, Poole T, Ryan NS, et al. Serum neurofilament light in familial Alzheimer disease: a marker of early neurodegeneration. Neurology. 2017;89(21):2167-75.
- Kovacs GG, Andreasson U, Liman V, et al. Plasma and cerebrospinal fluid tau and neurofilament concentrations in rapidly progressive neurological syndromes: a neuropathology-based cohort. Eur J Neurol. 2017;24(11):1326-e77.
- Rohrer JD, Woollacott IO, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87(13):1329-36.
- Hansson O, Janelidze S, Hall S, et al; Swedish BioFINDER study. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88(10):930-7.
- Feneberg E, Oeckl P, Steinacker P, et al. Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology. 2018;90(19):e22-30.
- Verde F, Steinacker P, Weishaupt JH, et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(2):157-64.
- Skillbäck T, Mattsson N, Blennow K, et al. Cerebrospinal fluid neurofilament light concentration in motor neuron disease and frontotemporal dementia predicts survival. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(5–6):397-403.
- Blennow K, Brody DL, Kochanek PM, et al. Traumatic brain injuries. Nat Rev Dis Primers. 2016;2:16084.
- Shahim P, Gren M, Liman V, et al. Serum neurofilament light protein predicts clinical outcome in traumatic brain injury. Sci Rep. 2016;6:36791.
- Shahim P, Zetterberg H, Tegner Y, et al. Serum neurofilament light as a biomarker for mild traumatic brain injury in contact sports. Neurol-ogy. 2017;88(19):1788-94.
- Moseby-Knappe M, Mattsson N, Nielsen N, et al. Serum neurofilament light chain for prognosis of outcome after cardiac arrest. JAMA Neurol. 2019;76(1):64-71.
- Zetterberg H, Wilson D, Andreasson U, et al. Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther. 2013;5(2):9.
- Mattsson N, Zetterberg H, Janelidze S, et al; ADNI Investigators. Plasma tau in Alzheimer disease. Neurology. 2016;87(17):1827-35.
- Gonzalez-Ortiz F, Karikari TK, Bentivenga GM, et al. Levels of plasma brain-derived tau and p-tau181 in Alzheimer’s disease and rapidly progressive dementias. Alzheimers Dement. 2023;20(1):745-51.
- Gonzalez-Ortiz F, Turton M, Kac PR, et al. Brain-derived tau: a novel blood-based biomarker for Alzheimer’s disease-type neurodegeneration. Brain. 2023;146(3):1152-65.
- Gonzalez-Ortiz F, Kirsebom BE, Contador J, et al. Plasma brain-derived tau is an amyloid-associated neurodegeneration biomarker in Alzheimer’s disease. Nat Commun. 2024;15(1):2908.
- Malmeström C, Haghighi S, Rosengren L, et al. Neurofilament light protein and glial fibrillary acidic protein as biological markers in MS. Neurology. 2003;61(12):1720-5.
- Dotevall L, Hagberg L, Karlsson JE, et al. Astroglial and neuronal proteins in cerebrospinal fluid as markers of CNS involvement in Lyme neuroborreliosis. Eur J Neurol. 1999;6(29):169-78.
- Wallin A, Blennow K, Rosengren LE. Glial fibrillary acidic protein in the cerebrospinal fluid of patients with dementia. Dementia. 1996;7(5):267-72.
- Nylén K, Csajbok LZ, Ost M, et al. Serum glial fibrillary acidic protein is related to focal brain injury and outcome after aneurysmal sub-arachnoid hemorrhage. Stroke. 2007;38(5):1489-94.
- Nylén K, Ost M, Csajbok LZ, et al. Increased serum-GFAP in patients with severe traumatic brain injury is related to outcome. J Neurol Sci. 2006;240(1–2):85-91.
- Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid-beta but not tau pathology in Alzheimer’s disease. Brain. 2021;144(11):3505-16.
- Benedet AL, Milà-Alomà M, Vrillon A, et al; Translational Biomarkers in Aging and Dementia (TRIAD) study; Alzheimer’s and Families (ALFA) study; BioCogBank Paris Lariboisière cohort. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471-83.
- Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl Psychiatry. 2021;11(1):27.
- Benussi A, Ashton NJ, Karikari TK, et al. Serum glial fibrillary acidic protein (GFAP) is a marker of disease severity in frontotemporal lobar degeneration. J Alzheimers Dis. 2020;77(3):1129-41.
- Veje M, Griška V, Pakalnienė J, et al. Serum and cerebrospinal fluid brain damage markers neurofilament light and glial fibrillary acidic protein correlate with tick-borne encephalitis disease severity – a multicentre study on Lithuanian and Swedish patients. Eur J Neurol. 2023;30(10):3182-9.
- Hansson O, Edelmayer RM, Boxer AL, et al. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimers Dement. 2022;18(12):2669-86.
Summary
Technical developments have paved the way for the development of ultrasensitive analytical methods that allow for precise quantification of brain-specific proteins in blood samples. Plasma levels of amyloid β, specifically the Aβ42/40 ratio, are reduced in Alzheimer’s disease (AD) and show concordance with brain amyloidosis assessed by PET, but the overlap with normal elderly may be too large for reliable use in clinical applications. Plasma phosphorylated tau (P-tau), especially a variant called P-tau217, is markedly increased in the early symptomatic stages of AD but remains normal in other neurodegenerative disorders.
Total tau (T-tau) is measurable in blood and shows most promise as a biomarker for acute neuronal injury (e.g. acute traumatic or hypoxic brain injury), where T-tau shows a fast and dramatic increase but does not work well as an AD biomarker due to contributions to blood levels from peripheral tissues. Instead, a novel method for tau protein produced only in the CNS called brain-derived tau (BD-tau) shows promise as a biomarker for AD-type neurodegeneration. Neurofilament light (NFL) levels in blood correlate tightly with levels in CSF and reflect axonal injury irrespective of the underlying cause.
Increased blood NFL concentration is found in several neurodegenerative disorders, including AD, but even more so in disorders such as motor neuron disease and frontotemporal dementia. Glial fibrillary acidic protein (GFAP) is expressed with activation of astrocytes, and is mildly increased in AD, but is also very high also in acute brain disorders. These blood tests show promise as tools to identify AD pathophysiology in the first assessment of patients with early cognitive symptoms, also in primary care, to guide clinical management and possible admission to the specialist clinic. A two-step model will result in a very high accuracy to either predict or exclude brain amyloidosis of the Alzheimer type.
Den centrala delen av er text är väl denna: ”Detta är viktigt eftersom det utöver psykologiska konsekvenser av en felaktig diagnos, till exempel baserad på ett falskt positivt resultat från ett blodtest, också finns andra aspekter på ett test som appliceras på stora patientgrupper för att ställa diagnos och initiera behandling. Det gäller både kostnader och, inte minst, potentiella medicinska risker med behandling för en sjukdom som patienten inte har.”
Så länge vi inte har en effektiv och någorlunda biverkningsfri behandling av denna svåra sjukdom, leder tidig diagnostik väl bara till elände för den som får en blodbaserad diagnos att leva med i ett liv som kanske inte blir vare sig kortare eller mer kopplat till senare Alzheimers sjukdom.
Även om de falskt positiva inte är mer än några procent av de undersökta leder ett sådant resultat för många människor sannolikt bara till sämre återstående livskvalitet. Som allmänläkare är det inte så sällan man får möta människor som farit illa av onödiga diagnoser baserade på osäkra tester för olika medicinska diagnoser. Det är en etisk baksida av tidig men osäker diagnostik.
Den dag den här sortens bloddiagnostik blir utbredd eller rent av allmän, kommer människor som får sin senare del av livet förgiftat av att leva med en föreställning om en diagnos som aldrig kom, kunna bli många.