Autoimmun nodopati är en neurologisk behandlingsbar sjukdom med nodala–paranodala antikroppar i serum mot celladhesionsmolekyler i Ranviers noder i perifera nerver.
Autoimmun nodopati betraktas inte längre som en variant av kronisk inflammatorisk demyeliniserande polyradikuloneuropati (CIDP) eller »seropositiv« CIDP, utan är en egen entitet.
Den mest aggressiva formen är pan-neurofascin autoimmun nodopati med IgG-antikroppar mot neurofascin-155 och neurofascin-186.
B-cellshämmande terapi (rituximab) är vanligen effektiv och prognosen kan vara god, även efter mycket långvarig intensivvård.
Specialiserad intensivvårdsrehabilitering kan bidra till autonomi och ett snabbare tillfrisknande.
Upptäckten av nodala–paranodala antikroppar i blod har under det senaste decenniet lett till identifiering av den nya neurologiska sjukdomen autoimmun nodopati [1, 2]. Sjukdomen har en karakteristisk fenotyp med distinkt immunpatologi och behandlingssvar. Tidigare har den rapporterats hos 5–10 procent av patienter med kliniska och neurofysiologiska kriterier för kronisk inflammatorisk demyeliniserande polyradikuloneuropati (CIDP) [2], men enligt de senaste CIDP-riktlinjerna från 2021 betraktas autoimmun nodopati numera som en egen entitet [3]. Korrekt diagnostik och behandling kan ge fullständig klinisk återhämtning, trots initiala tecken på axonala skador [1, 4]. Till skillnad från Guillain–Barrés syndrom (GBS) och CIDP svarar autoimmun nodopati sällan på intravenösa immunglobuliner (IVIg), som brukar ges vid inflammatoriska neuropatier, men ofta bra på B-cellshämmande terapi såsom rituximab [1, 5, 6].
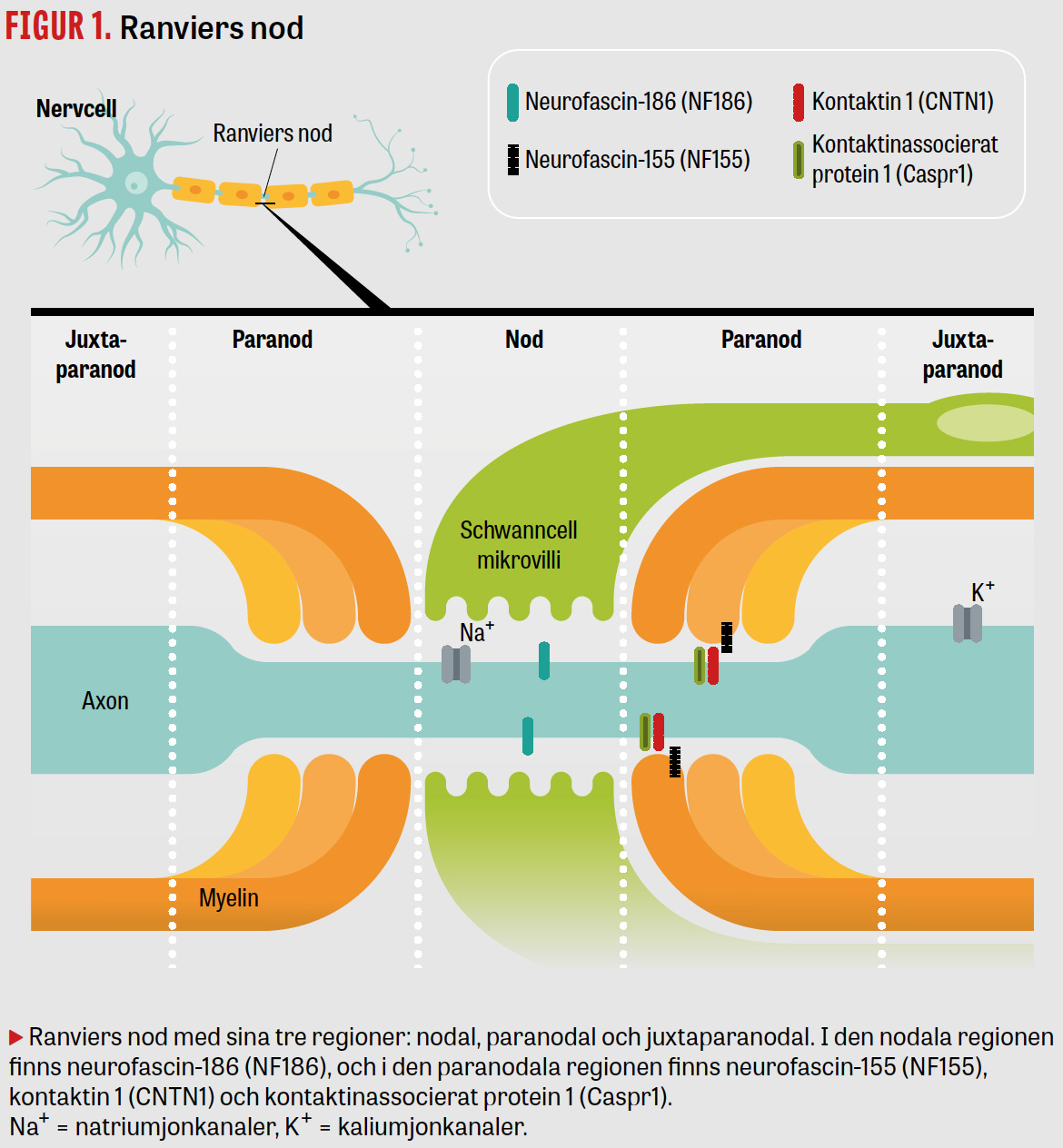
Vid autoimmun nodopati riktar sig nodala–paranodala antikroppar mot celladhesionsmolekyler i Ranviers noder i perifera nerver, neurofascin-155 (NF155), neurofascin-186 (NF186), kontaktin 1 (CNTN1) och kontaktinassocierat protein 1 (Caspr1) samt den nyligen beskrivna leucinrikt gliominaktiverat protein-4 (LGI-4) [1, 2, 7-11] (Figur 1). Detta orsakar en elektrofysiologisk blockering av nervkonduktion och leder obehandlat till destruktion av noden [1, 12, 13]. De nodala–paranodala antikropparna är av IgG-klass [5, 14]. IgM-antikroppar har beskrivits, men deras kliniska signifikans är oklar [15, 16]. Den vanligaste förekommande IgG-subklassen är IgG4, men även IgG3, IgG1 och IgG2 samt subklassbyte har rapporterats, vilket kan förklara olika behandlingssvar och bör beaktas vid val av behandling [5, 6, 9, 10, 17, 18]. Till skillnad från andra immunglobuliner aktiverar IgG4 inte cellulär eller komplementmedierad immunitet, vilket gör att IVIg och övriga konventionella immunterapier inte får avsedd effekt [5, 6]. I stället blockerar IgG4-antikroppar protein–proteininteraktion och signaltransduktion [1, 5, 14]. Neurofysiologiskt ses fynd som liknar demyeliniserande neuropati med låg nervledningshastighet, förlängd distal latens, förlängd F-svarslatens och konduktionsblockering (utan temporal dispersion) samt tidig förekomst av axonala skador och akut denervation [1, 2, 4, 19].
Autoimmun nodopati debuterar i form av en akut eller subakut sensorimotorisk neuropati som drabbar samtliga extremiteter med ett relativt aggressivt förlopp. Tetraplegi, kranialnervspåverkan, dysautonomi, respiratorisk svikt och glomerulonefrit kännetecknar den mest aggressiva formen kallad pan-neurofascin (pan-NF) autoimmun nodopati, medan uttalad ataxi, postural tremor, neuropatisk smärta och dåligt behandlingssvar på första linjens immunterapi brukar vara framträdande vid autoimmun nodopati med kroniskt förlopp [1, 2, 12, 14].
Vi beskriver här två patienter med pan-neurofascin autoimmun nodopati och redovisar deras sjukdomsutveckling, utredning, behandling och prognos.
Fall 1
En 57-årig tidigare frisk man sökte akut med 5 dagars anamnes på bilateral proximal muskelsvaghet i armarna i slutet av maj 2022. Inom några dagar tillkom lätt muskelsvaghet i benen (proximalt mer än distalt) och stickningar i händerna motsvarande 2 av 6 poäng på GBS disability scale (GBS-ds). Reflexer var svårutlösta och Babinskis tecken saknades. Likvoranalys visade ingen pleocytos, lätt stegrad albuminkvot (11,7; åldersreferens <9) och NFL (neurofilament light) (970 ng/l; åldersreferens <890 ng/l). Elektroneurografi visade tecken på demyelinisering i motoriska nerver. Behandling med IVIg 2 g/kg (totaldos) inleddes på misstanke om GBS, varvid armfunktionen förbättrades något och patienten skrevs ut till rehabiliteringsklinik. Tre dagar senare lades han åter in på neurologkliniken på grund av försämring med tilltagande tetrapares. Andningspåverkan tillkom, och han flyttades till intensivvårdsavdelning (Iva) 18 dagar efter symtomdebut. Han var då vaken och klar, hade en uttalad tetraplegi motsvarande GBS-ds 4 poäng, ensidig perifer facialispares och svalgpares, generell areflexi och känselnedsättning distalt i samtliga extremiteter samt var cirkulatoriskt instabil. Andningsproblemen ledde snabbt till intubation och ventilatorbehandling (GBS-ds 5 poäng). IVIg-behandlingen upprepades på grund av misstanke om en GBS-terapirelaterad fluktuation. Förnyad elektroneurografi visade tillkomst av konduktionsblockeringar som tolkades tala för GBS. Magnetkameraundersökning av hjärnan var normal, medan MR rygg visade kontrastladdande lumbosakrala nervrötter och conus medullaris.
Efter 46 dagar på Iva flyttades patienten till Remeo, en klinik för intensivvårdsrehabilitering för patienter med långvarig kritisk sjukdom. Den gradvisa försämringen fortsatte, och drygt 1 månad senare, 3 månader efter symtomdebut, tillkom bilateral perifer facialispares, tungpares, påverkad ögonmotorik och nackmuskulatur. Ny lumbalpunktion visade kraftigt förhöjd albuminkvot (49,6) och NFL (6 200 ng/l). En förnyad neurofysiologisk undersökning (4 månader efter symtomdebuten) visade en utbredd sensorimotorisk demyeliniserande och axonal neuropati samt denervation av flertalet undersökta extremitetsmuskler. Patienten var vid denna tid fortsatt tetraplegisk och respiratorberoende (GBS-ds 5 poäng). Underhållsbehandling med IVIg 1 g/kg var 3:e vecka påbörjades på misstanke om akutdebuterande CIDP (A-CIDP), men förbättring uteblev. I stället sågs en försämring av kranialnervsfunktioner de följande månaderna. Även den autonoma instabiliteten förvärrades med spontant kraftigt fluktuerande blodtryck samt uttalad ortostatism. Åtskilliga såväl blodtryckshöjande som -sänkande preparat provades. Bäst fungerade midodrin och enteral volym för att förebygga blodtrycksfall samt Trendelenburgs läge (sänkt huvudända/höjd fotända hos liggande patient) vid akuta tryckfall. Högre tryck förhindrades med lågdos angiotensin II-receptorblockerare och betablockad i kombination.
I december 2022, drygt 6 månader efter symtomdebut, byttes behandlingen mot intravenöst metylprednisolon under en fortsatt misstanke om CIDP, samtidigt som blodprov för nodala–paranodala antikroppar beställdes. Snart därefter kunde diagnosen pan-neurofascin autoimmun nodopati ställas i och med påvisande av IgG-antikroppar i serum mot såväl NF155 som NF186. Antikroppssvaret validerades på ett externt laboratorium i Tyskland, och kompletterande analys visade antikroppstiter på 1:500 av subklass IgG4. Behandling med rituximab (totaldos 2 000 mg) gavs, 7 månader efter symtomdebuten. Patienten var då fortsatt tetraplegisk och beroende av invasiv ventilatorbehandling via trakeostomi. NFL i serum var kraftigt förhöjt (260 ng/l; åldersreferens <19 ng/l).
Ungefär 1 månad efter behandling med rituximab, under fortsatt vård på Remeo, började patienten förbättras motoriskt. Tiden med talventil och högflödesbehandling utan ventilator [20] ökades de följande månaderna i takt med att muskelkraften återkom. I april 2023, 11 månader efter symtomdebuten, kunde ventilatorstödet avslutas. Den totala ventilatorbehandlingstiden blev 10 månader. Patienten dekanylerades i maj 2023. Drygt 1 vecka därefter skrevs han ut till rehabiliteringsklinik sittande i rullstol (GBS-ds 4 poäng). Därifrån skrevs han ut som gångare (GBS-ds 3 poäng) till hemmet, drygt 14 månader efter symtomdebuten. I november 2023 (1,5 år efter symtomdebut och 11 månader efter rituximabbehandlingen) kunde han gå utan stöd (GBS-ds 2 poäng), hade inga kranialnervspareser, var fortsatt något svag i händer och fötter samt rapporterade kognitiv fatigue. Han började arbeta deltid i januari 2024.
Efter rituximabbehandlingen normaliserades nivåerna av NFL i serum och titern av anti-pan-NF-antikroppar sjönk parallellt med den kliniska förbättringen och var till slut inte längre detekterbara.
Fall 2
En 49-årig tidigare frisk kvinna sökte akut med 3 dagars anamnes på svaghet i händerna i mitten av maj 2023. Inom 4 dagar tillkom bilateral proximal muskelsvaghet i armar och sensoriska retningssymtom i händer, fötter och läppar. Hon hade svårutlösta reflexer, och Babinskis tecken saknades. Den följande veckan tilltog muskelsvagheten i armarna och bilateral proximal muskelsvaghet tillkom i benen (GBS-ds 2 poäng). Likvoranalys visade lätt–måttligt förhöjd albuminkvot (11,8; åldersreferens <7), ingen pleocytos eller oligoklonala band, samt normalt NFL. Däremot var NFL lätt förhöjt i serum, medan övriga blodprov inklusive elfores var invändningsfria. Elektroneurografi visade tecken på lätt demyelinisering av motoriska nerver. På misstanke om GBS gavs IVIg 2 g/kg (totaldos) och patienten skrevs ut till rehabiliteringsklinik. Hon kom dock tillbaka 2 dagar senare (15 dagar efter symtomdebut) på grund av försämring med nytillkommen och tilltagande tetrapares (armar mer än ben) motsvarande GBS-ds 3 poäng. Ny elektroneurografi var oförändrad. På misstanke om GBS-terapirelaterad fluktuation alternativt A-CIDP gavs IVIg igen, vilket ledde till stabilisering och möjligen förbättrad handfunktion. Samtidigt beställdes analys av nodala–paranodala antikroppar, och patienten skrevs ånyo ut till rehabiliteringskliniken. Redan dagen därpå blev hon återinlagd på neurologkliniken efter ytterligare försämring. Hon var nu, 4 veckor efter symtomdebuten, närmast tetraplegisk med generell areflexi, svaghet i nacken och en svag hoststöt (GBS-ds 4 poäng). Ny lumbalpunktion visade oförändrad albuminkvot. NFL var lätt stegrat i likvor (1 180 ng/l; åldersreferens <890 ng/l) och serum (26 ng/l; åldersreferens <19 ng/l). NFL i serum steg senare kraftigt till 180 ng/l. Förnyad elektroneurografi visade tillkomst av konduktionsblockeringar, och behandling med intravenöst metylprednisolon och plasmaferes inleddes på misstanke om A-CIDP. Trots detta fortsatte patienten att försämras med tillkomst av respiratorisk svikt och autonom instabilitet, varför hon intuberades och flyttades till Iva (GBS-ds 5 poäng) drygt 1 månad efter symtomdebut. Hon hade då en uttalad tetraplegi med generell areflexi och svårbehandlade neuropatiska smärtor, men var efter trakeotomi vaken och klar. Nytillkommen diplopi föranledde MR hjärna, som var normal. Uttalad autonom instabilitet med kraftiga blodtrycksvariationer, takyarytmier samt tillkomst av nefrotiskt syndrom komplicerade vården. Utredning av anemi, Hb 61 g/l som lägst, påvisade inte någon blödningskälla. Svar på nodala–paranodala antikroppar validerades också på ett externt laboratorium i Tyskland och visade IgG-antikroppar mot NF155 och NF186 med antikroppstiter 1:200 av subklassen IgG4. Diagnosen pan-neurofascin autoimmun nodopati ställdes och rituximabbehandling (totaldos 2 000 mg) gavs, 1,5 månader efter symtomdebuten. Efter drygt 4 veckor på Iva flyttades patienten till Remeo för intensivvårdsrehabilitering.
På Remeo kunde tiden ur ventilator, med talventil och högflödesbehandling, relativt snabbt ökas från några minuter åt gången tills hon 2 veckor senare var helt utan ventilatorstöd. Svalgrehabilitering genomfördes samtidigt, och hon dekanylerades 2,5 månader efter symtomdebut. Muskelkraften återkom successivt, och vid utskrivning till rehabiliteringsklinik 1 månad efter rituximabbehandlingen kunde hon gå utan stöd (GBS-ds 2 poäng). I slutet av augusti skrevs patienten ut till hemmet, 3,5 månader efter symtomdebuten. I december noterades en nästan fullständig återhämtning förutom lätt nedsatt handstyrka (handdynamometer höger 29 kg, vänster 28 kg), men patienten rapporterade kognitiv fatigue. Hon började arbeta deltid i januari 2024.
Titern av anti-pan-NF-antikroppar och NFL i serum sjönk efter rituximabbehandlingen parallellt med den kliniska förbättringen. I oktober 2023, drygt 5 månader efter behandlingen, var antikropparna inte detekterbara och NFL i serum normaliserat. Hb normaliserades också, och det nefrotiska syndromet gick i regress.
Intensivvårdsaspekter
Modern intensivvård är mycket effektiv med möjlighet till avancerad monitorering och snabba, livräddande åtgärder. Enligt Svenska intensivvårdsregistret överlever 92 procent av patienterna trots mycket svår sjukdom och skrivs ut från Iva efter en genomsnittlig vårdtid på 2,5 dagar. Att i en sådan verksamhet samtidigt hantera vårdförlopp på veckor eller månader med väsentligt annorlunda patientbehov är en stor utmaning. Specialiserade kliniker har därför öppnats i flera länder [21-23]. Patienternas problematik är ofta mycket komplex [24], men specialiserade kliniker rapporterar trots detta goda resultat [22, 23].
Intensivvårdsrehabilitering är ett lagarbete med läkare, omvårdnadsteam, fysioterapeut, arbetsterapeut, logoped och kurator samt patient och närstående. Det interprofessionella teamet tar tillsammans med patient och närstående fram en individuell rehabiliteringsplan för urträning ur ventilator, evakuering av luftvägssekret, träning av munnens och svalgets muskulatur för att kunna hantera saliv och dekanylera samt mer traditionell träning för rörlighet och autonomi. Utvärdering och behandling av grundsjukdomen tillsammans med andra ansvariga specialiteter är central. För patienter med möjlig pan-neurofascin autoimmun nodopati innebär det ett nära samarbete med specialkunnig neurolog, eftersom det initiala förloppet är svårt att skilja från GBS och A-CIDP. Kontinuitet i det interprofessionella teamet är nödvändig för effektiv intensivvårdsrehabilitering, men kan vara svår att uppnå i traditionell intensivvård där jourbelastningen är hög [21]. Noggrann cirkulationsmonitorering är viktig eftersom påverkan på det autonoma nervsystemet kan vara uttalad med livshotande arytmier och blodtrycksförändringar, spontant eller som reaktion på sugning i luftvägar, positionsändring eller farmakologisk intervention.
Sekundära komplikationer, till exempel infektioner, trycksår och delirium, måste förebyggas genom noggrann monitorering och omvårdnad. Aktiva dagar efter varje patients förmåga är också centralt, liksom dygnsrytm med dagsljus och ostörda nätter för god sömn och återhämtning. Psykologiskt stöd för att hålla modet uppe hos både patient och närstående, möjlighet att vistas utomhus och umgås samt individuellt anpassade hjälpmedel för kommunikation kan hjälpa både tillfrisknande och delaktighet. Med detta arbetssätt är överlevnaden vid intensivvårdsrehabilitering generellt hög, på Remeo 97 procent [22], och återhämtningen fortsätter även efter utskrivning. Det finns därför skäl att konsultera en specialiserad klinik vid längre intensivvårdstider oavsett grunddiagnos, och särskilt vid GBS-liknande tillstånd.
Diskussion och konklusion
Pan-neurofascin autoimmun nodopati är en viktig differentialdiagnos till GBS och A-CIDP. Det finns dock inga tydliga kliniska skillnader vid insjuknande, även om våra patienter initialt hade en uttalad proximal muskelsvaghet i övre extremiteter, innan paresen generaliserades till tetraplegi. Det vanligaste spridningsmönstret vid klassisk GBS är däremot en ascenderande distal till proximal paralys med debut i nedre extremiteter. Det som i tidigt skede (de 4 första veckorna) skiljer pan-neurofascin autoimmun nodopati från klassisk GBS är det otillräckliga och fluktuerande terapisvaret på IVIg-behandling. Progress efter 8:e sjukdomsveckan skiljer pan-neurofascin autoimmun nodopati från GBS-terapirelaterad fluktuation, medan det är ytterst ovanligt med respiratorisk påverkan och dysautonomi vid A-CIDP.
Patienter med GBS behöver sällan så långvarig ventilatorbehandling som vi här beskriver vid pan-neurofascin autoimmun nodopati. I en studie av 149 patienter med GBS var mediantiden med invasiv ventilatorbehandling 30 dagar (variationsvidd 1–180 dagar) [25]. Eftersom pan-neurofascin autoimmun nodopati vanligtvis är en monofasisk sjukdom som kan gå i spontan remission utan anti-B-cellshämmande terapi [26] är det möjligt att några av patienterna i den ovannämnda studien hade pan-neurofascin autoimmun nodopati. Det går inte att ange en säker skillnad mellan GBS och pan-neurofascin autoimmun nodopati vad gäller rimlig tid med ventilatorbehandling. Vi föreslår att man analyserar nodala–paranodala antikroppar hos alla patienter med GBS med tetrapares med eller utan invasivt ventilatorstöd som inte förbättras 2–3 veckor efter immunterapi. Vår rekommendation är sammanfattad i Fakta 1. Antikroppsanalys kan beställas via Klinisk immunologi vid Karolinska universitetssjukhuset i Stockholm, Wieslab Diagnostic Services i Malmö och Likvorlaboratoriet på neurologmottagningen vid Karolinska universitetssjukhuset. Om nodala antikroppar påvisas är det viktigt med subklass och titerbestämning. Detta prov bör tas innan behandling med rituximab inleds.
Pan-neurofascin autoimmun nodopati är en av de första neurologiska sjukdomarna där upprepade mätningar av biomarkörer i blodet (NFL, anti-pan-NF-antikroppstiter samt lymfocytanalys) kan användas för utvärdering av behandlingseffekt och sjukdomsförlopp (Fakta 2) [12, 17, 27]. NFL i serum (eller plasma) kan användas som ett mått på nervskada och återhämtning, titeranalys som ett mått på nivån av antikroppar, som förväntas sjunka vid klinisk förbättring, samt B-lymfocytprofil som ett immunologiskt mått på rituximabeffekt.
Olika infusionsprotokoll för rituximab har föreslagits vid autoimmun nodopati: 1) 375 mg/m2 veckovis i 3 veckor och 2) 2 doser à 1 000 mg med 2 veckors intervall följt av ytterligare 1 dos 6 månader senare [12, 17]. Våra patienter behandlades med totaldos à 2 000 mg fördelat på 2 behandlingstillfällen inom 1 månad och utan ytterligare senare doser. Utöver behandling med rituximab finns rapporter om övergående eller stabiliserande effekt av kortikosteroider, plasmaferes och cyklofosfamid, samt positiv effekt av bortezomib och daratumumab för de fall som inte svarat på rituximab [12, 28, 29]. Våra två patienter hade ingen eller övergående effekt av IVIg. Den svaga effekten av IVIg som ses hos vissa patienter med autoimmun nodopati kan bero på förekomst av komplementaktiverande IgG3-/IgG1-antikroppar i ett tidigt stadium av autoimmun nodopati, innan dessa antikroppar genomgår subklassbyte till komplementoberoende IgG4 (IVIg har en hämmande effekt på komplement) [30].
Sammanfattningsvis är pan-neurofascin autoimmun nodopati ett allvarligt tillstånd med potentiellt god prognos vid korrekt diagnostik och behandling, inkluderande intensivvårdsrehabilitering (Fakta 3). Det är viktigt med tidig diagnostik och att ha den goda prognosen i åtanke och inte ge upp i väntan på rituximabeffekten när patienter med diagnostiserad pan-neurofascin autoimmun nodopati behöver långvarig ventilatorbehandling och intensivvård.
Eva Sundman var tidigare forskningschef vid Remeo, Stockholm.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Analys av nodala–paranodala antikroppar
Analys av nodala–paranodala antikroppar bör beställas i följande situationer vid misstänkt GBS och A-CIDP [3, 12, 28, 31]:
- aggressiv och allvarlig GBS-fenotyp med tetrapares (motorisk > sensorisk) med kort tid till nadir, inkluderande höggradig funktionsnedsättning och/eller neuromuskulär respiratorisk svikt och autonom påverkan
- uteblivet eller otillräckligt svar på första linjens immunterapier, IVIg och plasmaferes i fall av GBS eller IVIg, plasmaferes och kortison i fall av CIDP
- återfall kort (1–2 veckor) efter initialt behandlingssvar
- uttalad sensorisk ataxi med tremor och/eller kranialnervspåverkan
- fenotyp enligt ovan i kombination med höga nivåer av NFL i serum och albuminkvot; dessa kan dock ligga lågt under de första 1–2 veckorna
- fenotyp enligt ovan i kombination med nytillkommet nefrotiskt syndrom och/eller nyupptäckt lymfoproliferativ sjukdom
Fakta 2. Biomarkörer för uppföljning
Patienten bör följas både kliniskt och med regelbunden provtagning. Vid klinisk uppföljning bör man använda sig av validerade skattningsskalor såsom GBS-ds (Guillain-Barrés syndrome disability scale), INCAT (Inflammatory neuropathy cause and treatment disability score) och RODS (Rasch-built overall disability scale). Samtliga är tillgängliga på snema.se. Vi rekommenderar också att man tar följande blodprov var 3:e–6:e månad under det första året och därefter i samråd med en neuromuskulärt kunnig neurolog:
- NFL i serum (eller plasma)
- Nodal–paranodal antikroppstiteranalys
- B-lymfocytanalys
Fakta 3. Specialiserad intensivvårdsrehabilitering
Specialiserad intensivvårdsrehabilitering är indicerad för patienter som behöver intensivvård eller intermediärvård en längre tid, såsom de med pan-neurofascin autoimmun nodopati. Vi rekommenderar bedömning av läkare med kompetens inom intensivvårdsrehabilitering senast 21 dagar efter början av intensivvård. Vården och rehabiliteringen bedrivs i interprofessionella team och fokuserar på
- att genom basal intensivvård (fortsätta att) symtomatiskt behandla varje sviktande organsystem
- att förebygga och undvika sekundära komplikationer, till exempel infektioner, sekretstagnation, delirium, tromboemboliska händelser, trycksår och nedsatt rörelseförmåga
- att genom kontinuitet och noggrann monitorering tidigt upptäcka sjukdomsförändringar och, som patientens strategiska partner i samråd med andra ansvariga specialiteter, vidta åtgärder tidigt
- att aktivt stärka patientens autonomi och rehabilitera grundläggande funktioner som andning, hostning, sväljning, kommunikation och rörelseförmåga efter patientens individuella förutsättningar
- att under en lång och mycket utsatt livsperiod stödja patienten och de närstående i att skapa livskvalitet och hopp, både för tiden på sjukhus och livet därefter.
Referenser
- Martín-Aguilar L, Lleixà C, Pascual-Goñi E. Autoimmune nodopathies, an emerging diagnostic category. Curr Opin Neurol. 2022;35(5):579-85.
- Fehmi J, Vale T, Keddie S, et al. Nodal and paranodal antibody-associated neuropathies. Pract Neurol. Epub 26 maj 2021. doi: 10.1136/practneurol-2021-002960
- Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force – Second revision. J Peripher Nerv Syst. 2021;26(3):242-68.
- Khadilkar SV, Kamat S, Patel R. Nodo-paranodopathies: concepts, clinical implications, and management. Ann Indian Acad Neurol. 2022;25(6):1001-8.
- Dalakas MC. IgG4-mediated neurologic autoimmunities: understanding the pathogenicity of IgG4, ineffectiveness of IVIg, and long-lasting benefits of anti-B cell therapies. Neurol Neuroimmunol Neuroinflamm. 2022;9(1):e1116.
- Appeltshauser L, Brunder AM, Heinius A, et al. Antiparanodal antibodies and IgG subclasses in acute autoimmune neuropathy. Neurol Neuroimmunol Neuroinflamm. 2020;7(5):e817.
- Pascual-Goñi E, Fehmi J, Lleixà C, et al. Antibodies to the Caspr1/contactin-1 complex in chronic inflammatory demyelinating polyradiculoneuropathy. Brain. 2021;144(4):1183-96.
- Liu B, Zhou L, Sun C, et al. Clinical profile of autoimmune nodopathy with anti-neurofascin 186 antibody. Ann Clin Transl Neurol. 2023;10(6):944-52.
- Stengel H, Vural A, Brunder AM, et al. Anti-pan-neurofascin IgG3 as a marker of fulminant autoimmune neuropathy. Neurol Neuroimmunol Neuroinflamm. 2019;6(5):e603.
- Fehmi J, Davies AJ, Walters J, et al. IgG1 pan-neurofascin antibodies identify a severe yet treatable neuropathy with a high mortality. J Neurol Neurosurg Psychiatry. 2021;92(10):1089-95.
- Zhang X, Kira JI, Ogata H et al. Anti-LGI4 antibody is a novel juxtaparanodal autoantibody for chronic inflammatory demyelinating polyneuropathy. Neurol Neuroimmunol Neuroinflamm. 2023;10(2):e200081.
- Appeltshauser L, Doppler K. Pan-neurofascin autoimmune nodopathy – a life-threatening, but reversible neuropathy. Curr Opin Neurol. 2023;36(5):394-401.
- Manso C, Querol L, Mekaouche M, et al. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain. 2016;139(Pt 6):1700-12.
- Gupta P, Mirman I, Shahar S, Dubey D. Growing spectrum of autoimmune nodopathies. Curr Neurol Neurosci Rep. 2023;23(5):201-12.
- Doppler K, Stengel H, Appeltshauser L, et al. Neurofascin-155 IgM autoantibodies in patients with inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2018;89(11):1145-51.
- Shelly S, Klein CJ, Dyck PJB, et al. Neurofascin-155 immunoglobulin subtypes: clinicopathologic associations and neurologic outcomes. Neurology. 2021;97(24):e2392-403.
- Appeltshauser L, Junghof H, Messinger J, et al. Anti-pan-neurofascin antibodies induce subclass-related complement activation and nodo-paranodal damage. Brain. 2023;146(5):1932-49.
- Harris RE, Atherton M, Naude JTW, et al. Antineurofascin IgG2-associated paediatric autoimmune nodopathy. Dev Med Child Neurol. 2023;65(8):1118-22.
- Kouton L, Boucraut J, Devaux J, et al. Electrophysiological features of chronic inflammatory demyelinating polyradiculoneuropathy associated with IgG4 antibodies targeting neurofascin 155 or contactin 1 glycoproteins. Clin Neurophysiol. 2020;131(4):921-7.
- Egbers PH, Sutt AL, Petersson JE, et al. High-flow via a tracheostomy tube and speaking valve during weaning from mechanical ventilation and tracheostomy. Acta Anaesthesiol Scand. 2023;67(10):1403-13.
- Rak KJ, Ashcraft LE, Kuza CC, et al. Effective care practices in patients receiving prolonged mechanical ventilation. An ethnographic study. Am J Respir Crit Care Med. 2020;201(7):823-31.
- Löfroth M, Petersson JE, Uusijärvi J, et al. Outcomes of prolonged intensive care and rehabilitation at a specialized multidisciplinary center in Sweden. Acta Anaesthesiol Scand. 2022;66(2):232-9.
- Mifsud Bonnici D, Sanctuary T, Warren A, et al. Prospective observational cohort study of patients with weaning failure admitted to a specialist weaning, rehabilitation and home mechanical ventilation centre. BMJ Open. 2016;6(3):e010025.
- Herridge MS, Azoulay É. Outcomes after critical illness. N Engl J Med. 2023;388(10):913-24.
- Walgaard C, Lingsma HF, van Doorn PA, et al. Tracheostomy or not: prediction of prolonged mechanical ventilation in Guillain-Barré syndrome. Neurocrit Care. 2017;26(1):6-13.
- Uncini A. Autoimmune nodo-paranodopathies 10 years later: clinical features, pathophysiology and treatment. J Peripher Nerv Syst. 2023:28(Suppl 3):S23-35.
- Martín-Aguilar L, Lleixà C, Pascual-Goñi E, et al. Clinical and laboratory features in anti-NF155 autoimmune nodopathy. Neurol Neuroimmunol Neuroinflamm. 2021;9(1):e1098.
- Fels M, Fisse AL, Schwake C, et al. Report of a fulminant anti-pan-neurofascin-associated neuropathy responsive to rituximab and bortezomib. J Peripher Nerv Syst. 2021;26(4):475-80.
- Scheibe F, Ostendorf L, Pruss H, et al. Daratumumab for treatment-refractory antibody-mediated diseases in neurology. Eur J Neurol. 2022;29(6):1847-54.
- Querol L, Delmont E, Lleixà C. The autoimmune vulnerability of the node of Ranvier. J Peripher Nerv Syst. 2023;28(Suppl 3):S12-22.
- Van Doorn PA, Van den Bergh PYK, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society Guideline on diagnosis and treatment of Guillain-Barré syndrome. Eur J Neurol. 2023;30(12):3646-74.
Summary
Autoimmune nodopathy (AN) is an emerging diagnostic category with antibodies targeting the peripheral node of Ranvier. Clinically, AN overlaps with Guillain-Barré syndrome (GBS) and acute-onset CIDP. However, the immunopathology of AN is different, hence AN is considered as a separate entity. Antibodies targeting all neurofascin isoforms are associated with a phenotype and immunopathology described as pan-neurofascin (panNF) AN. Here, we describe a clinical and serological work-up of two patients with anti-panNF IgG4 AN, as well as treatment response. Due to the high morbidity of panNF and its treatable nature, we suggest early antibody testing in severe cases of GBS, especially when a prolonged need for invasive ventilation occurs. Specialized intensive care rehabilitation promotes autonomy and recovery, and should be considered as an alternative to traditional ICU care.