ALS är en heterogent neurodegenerativ sjukdom som främst drabbar de övre och nedre motorneuronen, men även andra delar av hjärnan kan drabbas.
37 gener har blivit kopplade till ALS. De vanligaste i Norden är C9orf72HRE, SOD1 och TBK1.
SOD1 kan spela en roll i sjukdomsutvecklingen även när genen i sig inte är muterad.
Experimentella studier pekar mot att ALS kan vara en prionsjukdom.
Allt fler läkemedelsprövningar genomförs i världen, flera med svenska patienter.
Neurologins fader Jean-Martin Charcot beskrev för 150 år sedan amyotrofisk lateralskleros (ALS) som ett degenerativt tillstånd med smygande debut av fokal pares i först ett enskilt myotom och därefter spridning under veckor–månader till närliggande myotom med bevarad sensorik och autonoma funktioner [1]. Med tiden förlamas all skelettmuskulatur med undantag av analsfinkter- och ögonmuskulaturen. Tecken på skada på övre och nedre motorneuron är essentiellt för diagnosen och speglar de mest framträdande obduktionsfynden – muskelatrofi och sklerosering av de nedåtgående nervbanorna i ryggmärgen. Patienterna avled när till slut även respirationsmuskulaturen drabbades. Kunskapen om »La maladie de Charcot« har ökat dramatiskt på senare år, och numera betraktas ALS som ett kliniskt, patologiskt, epidemiologiskt och genetiskt heterogent syndrom där flera typer har koppling till andra neurodegenerativa sjukdomar som Parkinsons sjukdom och Huntingtons sjukdom. Patienter från hela Sverige har gjort viktiga bidrag till dessa framsteg, och vi anar nu möjligheter till effektiva terapier och även förebyggande behandling.
Epidemiologi
ALS har i de flesta befolkningsstudier uppvisat en incidens på 2–4 per 100 000 personår, en prevalens på 6–8 per 100 000 och en livstidsrisk som uppskattas till 1:450 för män och 1:600 för kvinnor [2, 3]. Symtomdebut sker i regel vid cirka 60 års ålder men med stor variation, och ca 10 procent är under 30 år vid insjuknandet. Statistiska analyser talar för att ALS främst drabbar en genetiskt predisponerad grupp [4]. Anhopning av patienter med ALS (kluster) på en liten ort eller inom ett yrke (t ex elektriker) har varit föremål för forskning och uppmärksamhet i medierna [5]. Med tillkomsten av longitudinella patientdatabaser med tillhörande biobanker och avancerade genetiska analyser kan vi nu visa att flera av dessa kluster har en genetisk bakgrund (se nedan) eller beror på bristfällig diagnostik. Fall av s k konjugal ALS, där kvinna och man som lever ihop båda drabbas av ALS, har rapporterats men är inte mera vanligt förekommande än förväntat givet livstidsrisken att drabbas av ALS. Sammanfattningsvis finns i dag inget som talar för att ALS är smittsam mellan människor.
I prospektiva kohortstudier (t ex NEUROEPIC med 517 000 personer från hela Europa som på 1980-talet lämnade uppgifter om livsstil, exponeringar och sjukhistorik och som 25–35 år senare har följts upp med sjukdomsincidens) har de viktigaste riskfaktorerna för ALS varit förekomst av främst ALS eller frontallobsdemens men även Parkinsons sjukdom, demenssjukdom och schizofreni i släkten, hög ålder, manligt kön, rökning och smal kroppskonstitution [3, 6, 7]. Kraftig fysisk aktivitet har diskuterats som riskfaktor [8, 9], men ett samband kunde inte alltid visas [10]. Glädjande tycks det också finnas faktorer som minskar risken: i några studier är högt intag av kaffe eller alkohol associerat med lägre risk att drabbas [11]. Särskilt intressant är att personer med högt BMI och/eller typ 2-diabetes tycks ha lägre risk att insjukna och bättre prognos än övrig befolkning [7].
Neurogenetisk revolution
I retrospektiva epidemiologiska studier rapporteras att 5–10 procent av patienterna med ALS har en närstående släkting med samma diagnos; detta betecknas som familjär ALS (fALS) och övriga fall som sporadisk ALS (sALS). Den pågående neurogenetiska revolutionen har dock helt förändrat denna bild: sedan 1993 har 37 gener identifierats som predisponerar för ALS, och fler är under utredning [12]. En översikt finns på Socialstyrelsens webbplats (https://www.socialstyrelsen.se/stod-i-arbetet/ovanliga-diagnoser/amyotrofisk-lateralskleros/). Vid jämförelse av DNA-profilen hos patienter med tidig respektive sen sjukdomsdebut samt kort respektive lång överlevnadstid har 5 gener identifierats som modifierande gener (»modifiers«), och bärarskap av olika varianter av dessa ger olika sjukdomsförlopp. De fem generna är CHGB, SMN2, PGC-1alfa, VEGF och EphA4. Upptäckten av modifierande gener ger insikt i sjukdomsmekanismen och kan öppna upp för nya behandlingsmöjligheter [13].
Sedan år 1993 har ALS-forskargruppen vid Umeå universitet samlat blodprov från patienter från hela Sverige, och omfattande molekylärbiologiska och genetiska analyser har utförts. Många patienter har bidragit, och stora släktträd har konstruerats där fyra distinkta nedärvningsmönster har identifierats:
- dominant med hög penetrans (minst 95 procent av anlagsbärare utvecklar sjukdom före 70 års ålder),
- dominant med nedsatt penetrans (inte alla anlagsbärare insjuknar kliniskt),
- recessivt (dvs en sjuk patient som bär på dubbla anlag från två friska men anlagsbärande föräldrar)
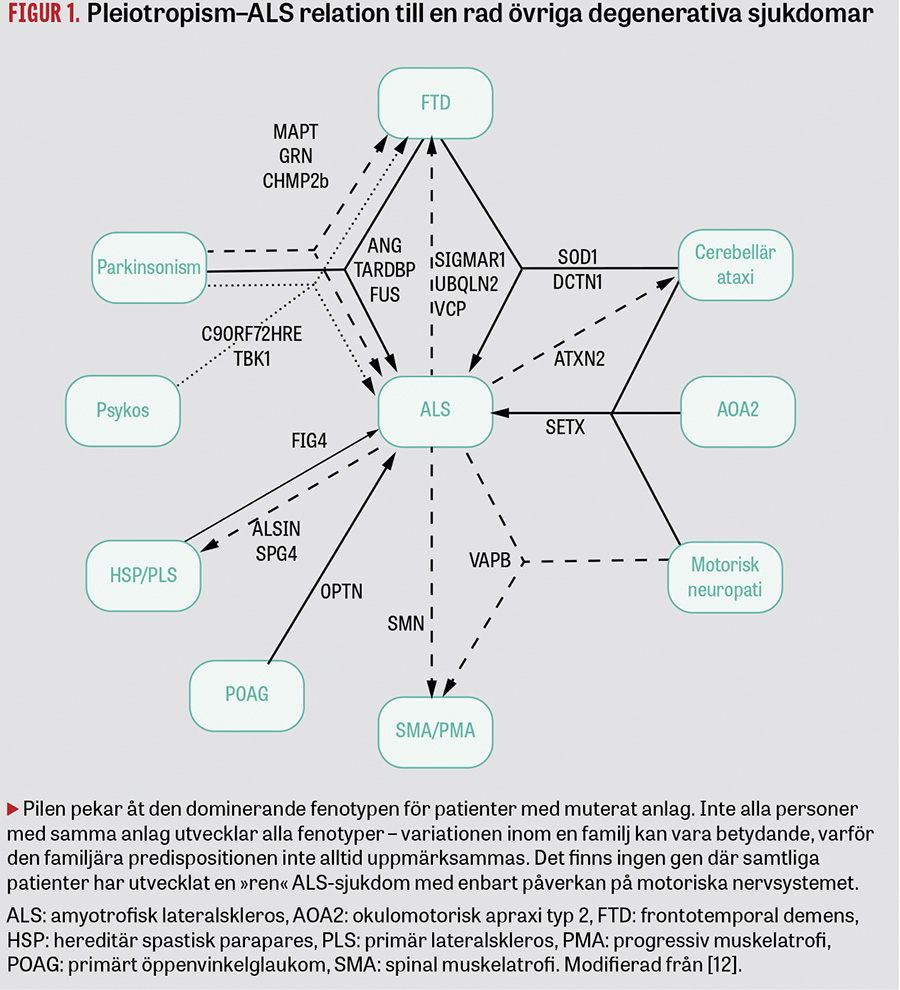
- dominant pleiotropiskt nedärvningsmönster (en och samma mutation kan ge upphov till olika fenotyper, t ex ALS, parkinsonism, frontallobsdemens [Figur 1].
Nedsatt penetrans och pleiotropism i kombination med ofullständig familjesjukdomshistorik samt bristfällig diagnostik antas vara orsaken till att 10–15 procent av sALS-patienter i DNA-analyser visar sig ha mutationer som tidigare är påvisade hos fALS-patienter [12]. Hos ca 18–25 procent av alla svenska ALS-patienter hittar vi en mutation i någon av de 37 kända ALS-generna. Att identifiera ärftliga sjukdomsanlag hos patienter med förmodad sporadisk/spontan ALS kan medföra en psykisk påfrestning för patienten och anhöriga och kan sätta läkaren i en besvärlig klinisk situation [14]. Genetisk rådgivning kan ibland behövas. Ett patologiskt DNA-svar öppnar för tidigare och säkrare diagnostik och för patientens medverkan i genspecifika terapistudier (se nedan).
De tre vanligaste sjukdomsgenerna vid ALS i Sverige är C9orf72HRE, SOD1 och TBK1; mutationer i de övriga generna är ovanliga. I C9orf72 finns en GGGGCC-hexanukleotidsekvens (HRE) som i vanliga fall upprepas ett fåtal gånger (<23 repetitioner anses som normalt), men som vid ALS och frontallobsdemens kan ha upp till 2 000 repetitioner. I en obduktionsstudie hittade vi inget tydligt samband mellan det totala antalet repetitioner i olika delar av CNS och fenotyperna ALS, ALS-demens eller frontallobsdemens. Anmärkningsvärt nog hade två av de 18 obducerade patienterna ett gränsvärde vad gäller antal repetitioner i leukocyter i blod men i varierande grad en kraftig expansion i olika delar av CNS [15].
C9orf72-proteinets normala funktion är ännu okänd, men den stora mängden extra DNA antas störa mRNA-metabolismen, resultera i bildning av repetitiva dipeptider (sådana kan påvisas i ryggmärgsvätskan) eller medföra haploinsufficiens. C9orf72HRE uppvisar s k pleiotropism, och bärare av mutationen kan drabbas av såväl ALS och/eller frontallobsdemens som parkinsonism eller schizofreni [12].
En av de viktigaste släkterna för upptäckten av C9orf72HRE som sjukdomsgen var en svensk släkt från Närke, där man på 1980-talet upptäckte en hög förekomst av ALS och frontallobsdemens med dominant nedärvningsmönster [16]. DNA-analyser 1998–99 från denna släkt och en liknande släkt i Västerbotten pekade mot ett nytt genlokus på kromosom 9p21 under hypotesen att ALS och frontallobsdemens i dessa familjer hade samma orsak [17]. Speciellt amerikanska neurologer motsatte sig denna teori, då ett etiologiskt samband med frontallobsdemens bröt mot Charcots dogm att kognitiv påverkan inte förekommer vid ALS [1]. Det var först efter att liknande amerikanska ALS/frontallobsdemens-släkter med koppling till 9p21 hade identifierats som C9orf72HRE-sjukdomsanlaget kunde identifieras år 2011 [18].
I dag vet vi att C9orf72HRE är den vanligaste kända orsaken till ALS och att den är sjukdomsorsak hos 10–14 procent av alla svenska patienter med ALS och 5–6 procent av patienter med frontallobsdemens [19, 20]. ALS med demens i olika grad ses ofta hos C9orf72HRE-bärande patienter, men ALS-patienter utan tydlig kognitiv påverkan efter flera års sjukdom förekommer också. Klinisk frontallobsfunktion hos ALS-patienter kan bedömas med ett 20 minuters ECAS-test utvecklat vid University of Edinburgh och validerat på svenska patienter och kontrollpersoner. Som tumregel har C9orf72HRE-patienter, och särskilt de med demens och parkinsonism, sämre prognos än patienter med »ren« ALS-sjukdom. Medan penetransen är hög i vissa släkter är den lägre i andra släkter, talande för att det finns ännu okända modifierande faktorer som påverkar om, när och i vilka delar av nervsystemet som degenerationen initialt ger symtom.
Mutationer i genen för superoxiddismutas 1 (SOD1) var den första identifierade orsaken till ALS, och sedan 1993 har 218 mutationer upptäckts, 14 hos patienter i Sverige [21, 22]. Cirka 6 procent av alla patienter bär på en SOD1-mutation. SOD1-genen kodar för ett litet cytoplasmatiskt enzym som neutraliserar fria syreradikaler och som finns i alla kroppens celler. Flertalet SOD1-mutationer medför nedsatt enzymaktivitet. År 1993 hittade vi mutationen D90A, som är den vanligaste SOD1-mutationen globalt och den näst vanligaste kända orsaken till ALS efter C9orf72HRE. Patienterna var homozygota för mutationen, och enzymaktiviteten var normal [23, 24]. Denna upptäckt, som initialt gjordes på 14 patienter, visade att ALS orsakas av en cytotoxisk effekt hos muterat SOD1 (»gain-of-function«) och inte av förlust av enzymaktivitet och en oförmåga att neutralisera fria radikaler, vilket var den förhärskande teorin på 90-talet.
ALS som SOD1-mallstyrd prionsjukdom
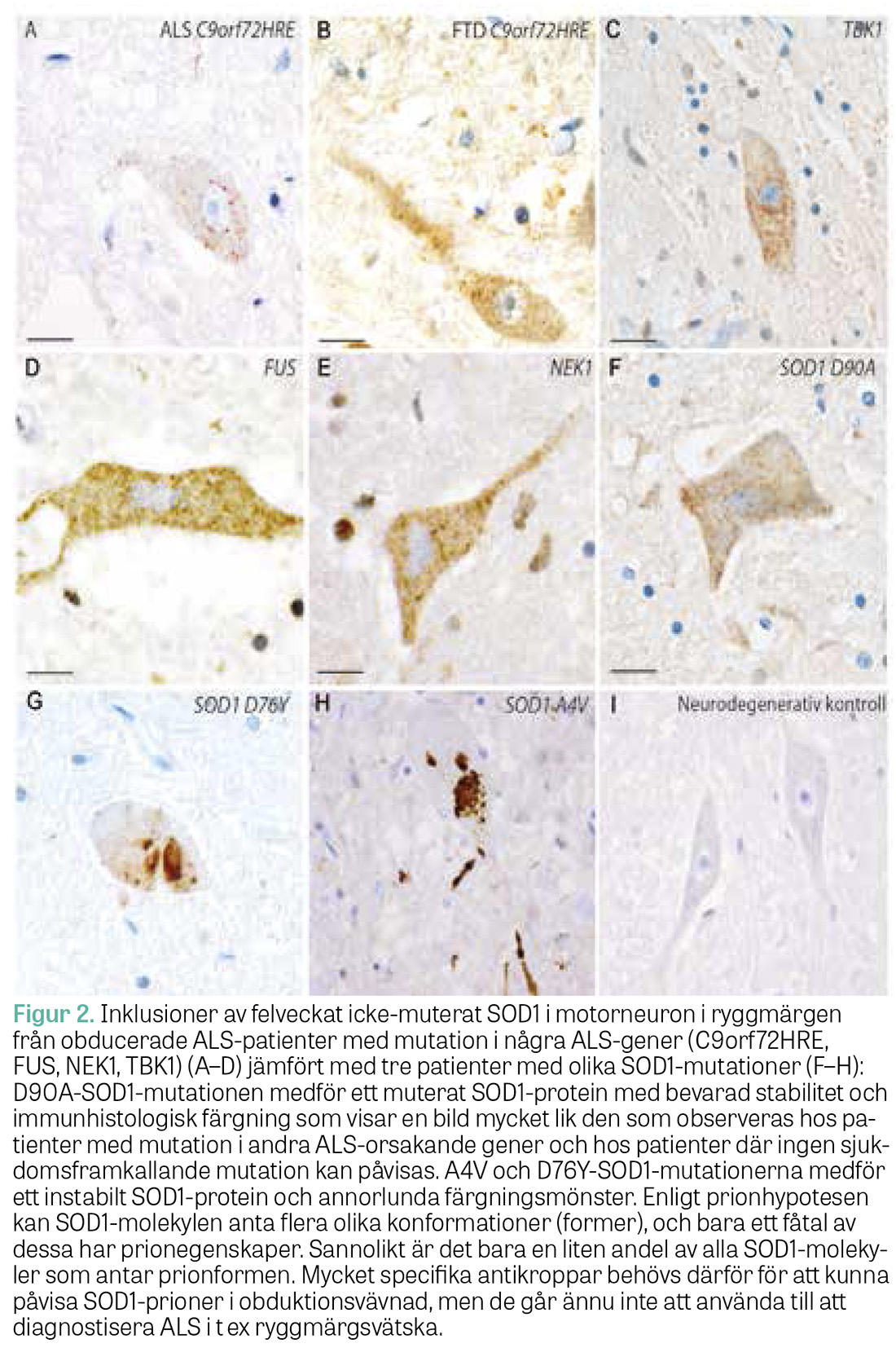
De neurogenetiska genombrotten har gett insikt i vilka proteiner som spelar roll vid ALS och associerade överlappande syndrom. De 37 identifierade sjukdomsgenerna kan indelas i 5 grupper efter proteinernas kända funktioner [12]. Medan vissa proteiner antas orsaka neurodegeneration genom förlorad funktion (t ex TBK1), antas flera verka via »gain-of-function« där det muterade proteinet har fått nya egenskaper. Bäst undersökt är SOD1-proteinet. I obduktionsstudier kan man med hjälp av antikroppar, specifika för icke-normalt veckat SOD1-protein, påvisa ansamlingar (s k inklusioner) av SOD1-protein i cytoplasman i motorneuron och i gliacellers kärnor [25, 26]. Inklusioner av felveckat SOD1 påvisas hos ALS-patienter med mutation i SOD1 men även hos patienter med mutation i andra ALS-associerade gener och patienter med sALS [27, 28] (Figur 2).
Överuttryck av muterat SOD1 och den normala SOD1-genen i möss resulterar i en åldersberoende fatal ALS-liknande sjukdom med bildning av SOD1-inklusioner främst i motoriska nervsystemet. Två strukturellt olika inklusionstyper av SOD1 har kunnat identifieras med användning av antikroppar specifika för olika epitoper i SOD1. Stereotaktisk injektion av preparationer av renat SOD1 i ryggmärgens framhorn på 100 dagar gamla möss medför 60–80 dagar senare utveckling av en fulminant ALS-sjukdom, som i enlighet med Charcots dogm börjar fokalt och sprider sig genom motoriska nervsystemet från den punkt där injektionen skedde. Samtidigt ses en mallstyrd bildning av SOD1-inklusioner med samma 3D-struktur som i preparationen som injicerades [29, 30]. Dessa inklusioner kan isoleras och injiceras i en ny mus med samma förödande resultat. Liknande resultat har vi uppnått vid injektion av renade SOD1-inklusioner från humana ALS-obduktioner [31]. Vi drar slutsatsen att SOD1-protein kan anta en specifik tredimensionell form som har prionliknande egenskaper.
Hittills har två sådana SOD1-prionstammar med olika struktur, spridningsmönster och prognos identifierats i ALS-möss. Det är sannolikt att liknande gäller hos människan [30]; däremot försvårar den låga halten av SOD1-aggregat hos en avliden ALS-patient, jämfört med en transgen mus med genöveruttryck, typbestämning av prionstammar hos patienter med dagens teknik. Viktigt att notera är att prionegenskaperna hos SOD1 verkar betydligt mindre stabila och infektiösa jämfört med »klassiska« prioner såsom vid Creutzfeldt–Jakobs sjukdom, vilket gör att ALS hos människor i praktiken fortfarande får anses som en icke-smittsam sjukdom. Orsaken till detta är de vitt skilda biokemiska egenskaperna hos SOD1-aggregat och aggregat av prionproteinet PrP.
Nya behandlingsstrategier
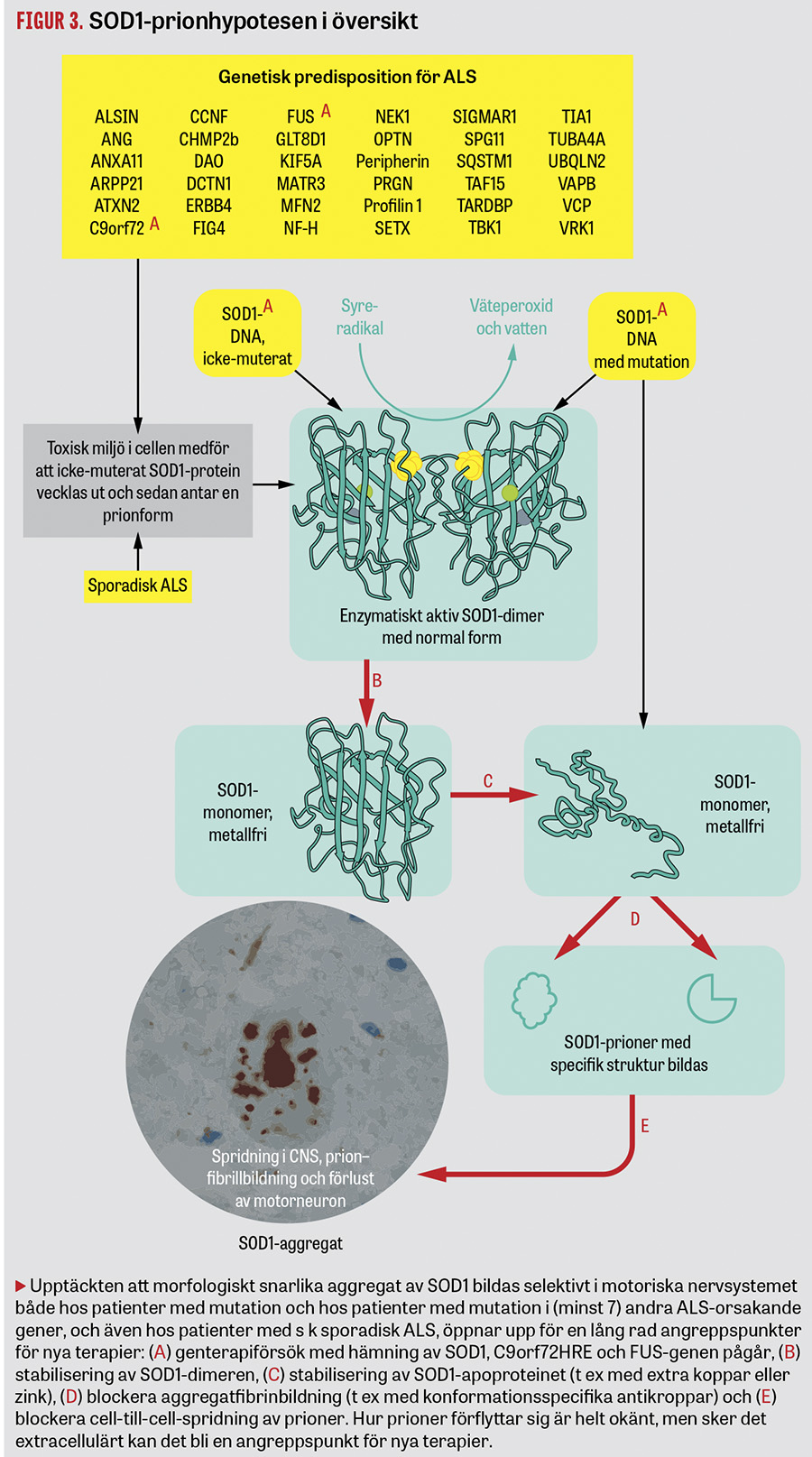
Med den kunskap som finns i dag tycks inklusioner av felveckat SOD1 finnas hos patienter med alla typer av ALS och många med frontallobsdemens. Vad som orsakar felveckning av SOD1 hos patienter med mutation i andra gener än SOD1 eller hos patienter med sALS är i dag okänt. Varför SOD1-prioner främst bildas i CNS (det finns två till tre gånger så mycket SOD1 i t ex njur- och levervävnad) och främst drabbar motoriska nervsystemet, och hur prionen fortplantas från cell till cell, är likaledes ännu okänt. Den nya kunskapen om den molekylära patogenesen öppnar upp för helt nya terapier inriktade på att minska den skadliga prionbildningen t ex genom att
- hämma syntesen av ytterligare SOD1-protein med genterapi
- stabilisera SOD1-proteinet så att det inte felveckas
- blockera bildningen av proteinaggregat
- öka nedbrytningen av proteinaggregat
- modulera immunsystemets respons på bildning och/eller proteinaggregaten
- blockera spridningen av prioner (Figur 3).
Flera sådana studier pågår eller planeras med svensk medverkan [32]. Under 2018–2019 har fler svenska patienter fått medverka i läkemedelsstudier än något tidigare år. Upptäckten av ALS-gener har sedan 1990-talet möjliggjort presymtomatiskt anlagsbärartest av patienters anhöriga [33]. Den nya upptäckten av konformationsspecifika SOD1-inklusioner som orsak till ALS kan möjliggöra utveckling av aktiv eller passiv immunterapi mot SOD1-aggregat. Dessutom pågår läkemedelsstudier med genuttryckshämmande läkemedel som »stänger av« sjukdomsanlagen.
Potentiella bindningar eller jävsförhållanden: Peter M Andersen är nationell huvudprövare för läkemedelsprövningarna REFALS (Orion Pharma), ORARIALS01 (Orphazyme A/S) och 233AS101 (Biogen). Karin Forsberg är medprövare för nämnda studier. Peter M Andersen ingår i rådgivningsgrupperna för ORARIALS- och 233AS101-studierna, är rådgivare för ALS Pharma, Biogen, Roche-Genentech och Voyager och ledamot av Hjärnfondens vetenskapliga nämnd (arvoderade uppdrag).
(uppdaterad 2020-03-16)
Referenser
- Charcot JM. Lectures on the diseases of the nervous system. Vol 2, serie 2 (red/övers Sigerson G). London: New Sydenham Society; 1881. p. 163-20.
- Forsgren L, Almay BG, Holmgren G, et al. Epidemiology of motor neuron disease in northern Sweden. Acta Neurol Scand. 1983;68(1):20-9.
- Haverkamp LJ, Appel, V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain. 1995;118(Pt 3):707-19.
- Neilson S, Robinson I, Nymoen EH. Longitudinal analysis of amyotrophic lateral sclerosis mortality in Norway, 1966-1989: evidence for a susceptible subpopulation. J Neurol Sci. 1994;122(2):148-54.
- Gunnarsson LG, Lindberg G, Söderfeldt B, et al. Amyotrophic lateral sclerosis in Sweden in relation to occupation. Acta Neurol Scand. 1991;83(6):394-8.
- Gallo V, Bueno-de-Mesquita HB, Vermeulen R, et al. Smoking and risk of amyotrophic lateral sclerosis: analysis of the EPIC cohort. Ann Neurol. 2009;65(4):378-85.
- Gallo V, Wark PA, Jenab M, et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: the EPIC cohort. Neurology. 2013;80(9):829-38.
- Chiò A, Benzi G, Dossena M, et al. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005;128(Pt 3):472-6.
- Huisman MH, Seelen M, de Jong SW, et al. Lifetime physical activity and the risk of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2013;84(9):976-81.
- Gallo V, Vanacore N, Bueno-de-Mesquita HB, et al. Physical activity and risk of ALS in a prospective cohort study. Eur J Epidemiol. 2016;31(3):255-66.
- de Jong SW, Huisman MH, Sutedja NA, et al. Smoking, alcohol consumption, and the risk of amyotrophic lateral sclerosis: a population-based study. Am J Epidemiol. 2012;176(3): 233-9.
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603-15.
- Corcia P, Ingre C, Blasco H, et al. Homozygous SMN2 deletion is a protective factor in the Swedish ALS population. Eur J Hum Genet. 2012;20(5):588-91.
- Eisen A, Mezei M, Stewart HG, et al. SOD1 gene mutations in ALS patients in British Columbia, Canada: clinical features, neurophysiology and ethical issues in management. Amyotroph Lateral Scler. 2008;9(2):108-19.
- Nordin A, Akimoto C, Wuolikainen A, et al. Extensive size variability of the GGGGCC- expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum Mol Genet. 2015;24(11):3133-42.
- Gunnarsson LG, Dahlbom K, Strandman E. Motor neuron disease and dementia reported among 13 members of a single family. Acta Neurol Scand. 1991;84(5):429-33.
- Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66(6):839-44.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245-56.
- Smith BN, Newhouse S, Shatunov A, et al. The C9ORF72 expansion mutation is a common cause of ALS+/–FTD in Europe and has a single founder. Eur J Hum Genet. 2013;21(1):102-8.
- Ferrari R, Hernandez DG, Nalls MA, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014;13(7):686-99.
- Rosen DR, Siddique T, Patterson D, et al. Mutations in CuZn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59-62.
- Andersen PM, Nilsson P, Keränen ML, et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain. 1997;120(Pt 10):1723-37.
- Andersen PM, Nilsson P, Ala-Hurula V, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet. 1995;10(1):61-6.
- Andersen PM, Forsgren L, Binzer M, et al. Autosomal recessive adult-onset ALS associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain. 1996;119(Pt 4):1153-72.
- Forsberg K, Jonsson PA, Andersen PM, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One. 2010;5(7):e11552.
- Bosco DA, Morfini G, Karabacak NM, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396-403.
- Forsberg K, Andersen PM, Marklund SL, et al. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121(5):623-34.
- Forsberg K, Graffmo K, Pakkenberg B, et al. Misfolded SOD1 inclusions in patients with mutations in C9ORF72 and other ALS/FTD-associated genes. J Neurol Neurosurg Psychiatry. 2019;90(8):861-9.
- Bidhendi EE, Bergh J, Zetterström P, et al. Inoculated SOD1 aggregate strain transmits amyotrophic lateral sclerosis with templated aggregation. J Clin Invest. 2016;126(6):2249-53.
- Ayers JI, Fromholt SE, O’Neal VM, et al. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 2016;131(1):103-14.
- Bidhendi EE, Bergh J, Zetterström P, et al. Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 2018;136(6):939-53.
- Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435-42.
- Benatar M, Stanislaw C, Reyes E, et al. Pre-symptomatic ALS genetic counseling and testing: experience and recommendations. Neurology. 2016;86(24):2295-302.