
Om 30 år kommer 250 000 personer i Sverige att ha en demenssjukdom.
Förklaringsmodellen för Alzheimers sjukdom är »amyloidkaskadhypotesen« där amyloid-β aggregerar, vilket skadar nervcellerna.
Den symtomatiska fasen börjar oftast med försämrat närminne.
Likvoranalyser och PET-kamera som detekterar amyloid-β och tau är tillgängliga för klinisk diagnostik.
Nuvarande terapi vid Alzheimers sjukdom baseras på två klasser av kognitionsförstärkande läkemedel: kolinesterashämmare och NMDA-receptorantagonister.
Den senaste utvecklingen av potentiella behandlingar för alzheimer är aktiv eller passiv immunterapi mot amyloid-β och tau, där några studier nyligen har uppvisat lovande effekter.
Alzheimers sjukdom är den absolut vanligaste demenssjukdomen; man beräknar att så många som 250 000 personer i Sverige har en demenssjukdom år 2050 [1]. Neuropatologiskt karakteriseras alzheimer av kombinationen amyloidos (plack av proteinet amyloid-β [Aβ]), tau-patologi i form av neurofibrillära nystan och neuropiltrådarsamt neurodegeneration (neuron- och synapsnedbrytning) [2, 3]. Det finns två former av plack vid alzheimer: »diffusa plack« som enbart består av löst aggregerad Aβ, och »kompakta plack« som har en kärna av hårt aggregerat Aβ, vilka är omgivna av skadade nervcellsutskott och aktiverade gliaceller.Aβ-aggregat och -plack påverkar även andra proteiner och fysiologiska processer, vilket leder till att det normala nervcellsproteinet tau fosforyleras och börjar aggregera till neurofibrillära nystan [4].
Den stora majoriteten av alla alzheimerfall är sporadiska, men det finns även en sällsynt (mindre än 1 procent av alla fall) ärftlig form kallad familjär Alzheimers sjukdom. De mutationer som leder till familjär autosomalt dominant Alzheimers sjukdom finns i amyloidprekursorproteinet (APP) eller i presenilin (PSEN)-generna.
Familjär Alzheimers sjukdom debuterar vanligen före 65 års ålder, medan hög ålder är den största riskfaktorn för sporadisk Alzheimers sjukdom, och risken att insjukna ökar betydligt efter 65 års ålder. Den viktigaste riskgenen är apolipoprotein E (APOE), där en variant kallad APOE ε4 ökar risken med upp till 10–12 gånger för homozygoter, och 3–4 gånger för heterozygoter [5].
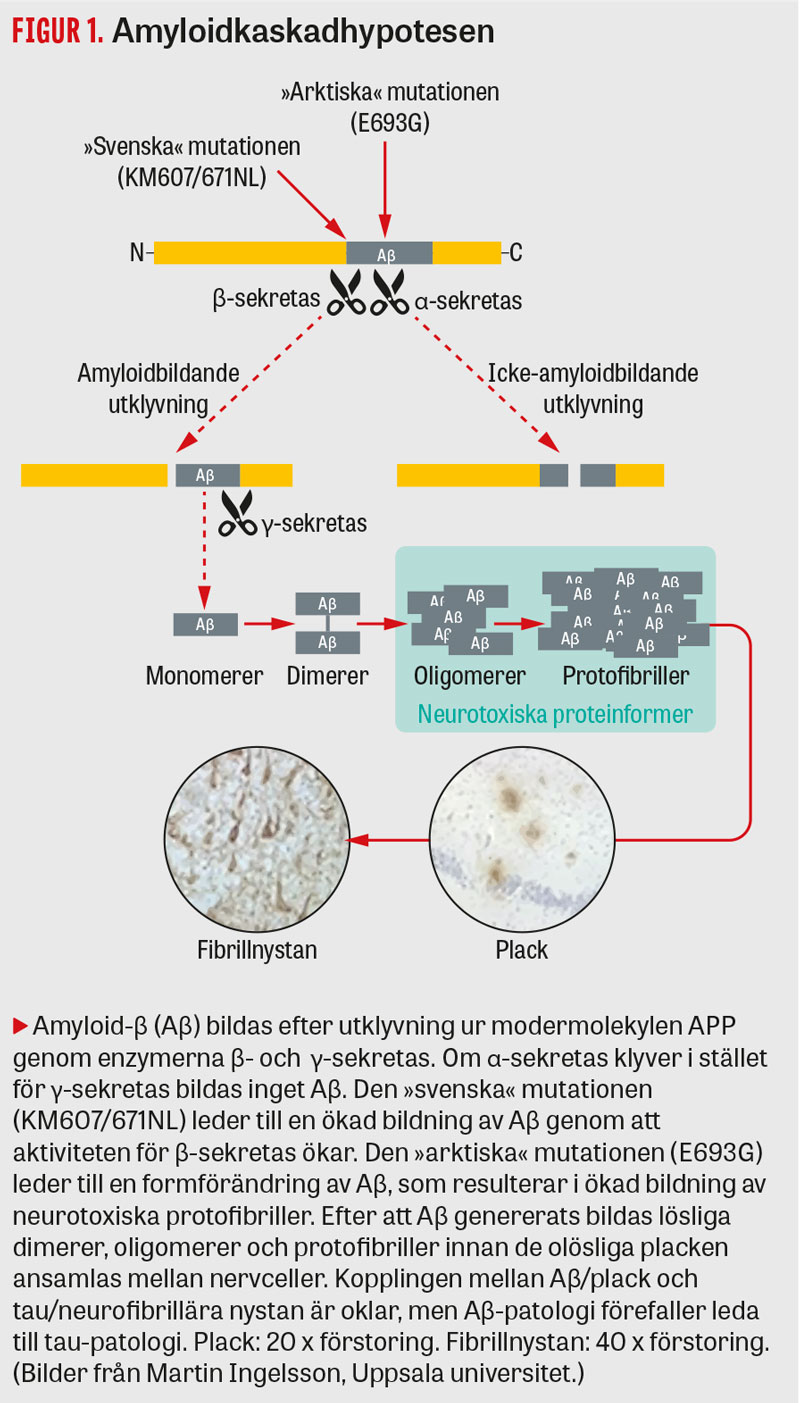
Den helt dominerande förklaringsmodellen för hur alzheimer uppkommer är den så kallade amyloidkaskadhypotesen, där ett fysiologiskt normalt protein i hjärnan (Aβ) börjar att aggregera, vilket antas försämra nervcellernas funktion (Figur 1). Orsaken till att Aβ börjar aggregera vid sporadisk alzheimer är inte känd, men sannolikt är det inte produktionen som är ökad utan nedbrytningen eller uttransporten från hjärnan (clearance) som är minskad [6]. Experimentella och kliniska observationer talar starkt för att det är Aβ som initierar sjukdomsprocessen.
Klinik
De kliniska kriterierna för Alzheimers sjukdom innefattar en kognitiv funktionsnedsättning som påverkar minst två av domänerna minne, resonemang och hantering av komplexa uppgifter, visuospatiala förmågor, språkfunktion samt personlighet och beteende. Det finns två huvudsakliga fenotyper av alzheimer: amnestisk, som är vanligast, och icke-amnestisk. De tydligaste symtomen är nedsatt inlärningsförmåga och svårigheter att återge nyligen tillägnad information, medan andra kognitiva förmågor påverkas mindre i början.
Alzheimers sjukdom har ett mångårigt förlopp med en långsam försämring över tid, men förloppet är högst varierande. I den tidiga fasen av sjukdomen, lindrig kognitiv svikt (mild cognitive impairment, MCI), uppvisar patienterna endast lätta minnesproblem, ofta tillsammans med beteendeförändringar som nedstämdhet och initiativlöshet, men utan störd ADL-förmåga. Senare progredierar symtomen till »demens«, vilket enligt DSM-IV-kriterierna definieras som minnesstörning i kombination med afasi, apraxi, agnosi eller nedsatt exekutiv förmåga av sådan grad att det påverkar den sociala förmågan eller arbetsförmågan. Basutredningen av alzheimer börjar med anamnes hos allmänläkare, datortomografi av hjärnan för att utesluta till exempel subduralhematom, Mini-mental test (MMT) för att gradera symtomen och blodprov såsom TSH för att utesluta sjukdomar som kan tänkas påverka kognitiv förmåga. Efter detta remitteras patienten till minnesmottagning där mera avancerade diagnostiska metoder (MRT, PET, EEG, likvoranalyser och neuropsykologisk testning) oftast används.
Den kliniska bilden vid alzheimer varierar och är ofta svår att skilja från andra neurodegenerativa sjukdomar, inte minst tidigt i förloppet. Efter en lång (flera decennier) preklinisk fas där patologin byggs upp debuterar sjukdomen oftast med försämrat episodiskt minne [7], men även icke-amnestiska former förekommer. Efter hand som den neurodegenerativa processen sprider sig från de mediala delarna av temporalloben över bakre temporoparietala kortex till hela hjärnbarken [8] utvecklas i typiska fall symtom i form av försämrad tankeförmåga, språkstörningar, försämrad rumsuppfattning, nedsatt praktisk förmåga och störningar i varseblivning.
De diagnostiska svårigheterna är störst vid den sent debuterande (>65 år) formen av sjukdomen. Anledningen till detta är att flertalet patienter inte bara har amyloid- och tau-patologi, utan också kombinationer av alzheimerförändringar med till exempel Lewykroppspatologi, TDP-43-utfällning, cerebrovaskulär sjukdom eller hippocampusskleros [9]. Baserat på likvor- och PET-biomarkörer som kan påvisa alzheimerpatologi hos levande patienter presenterade National Institute on Aging och Alzheimer’s Association nyligen en ny definition av Alzheimers sjukdom, som bygger på att patologin identifieras med biomarkörer enligt en så kallad A/T/N-klassificering, där A står för amyloidos, T för tau-patologi och N för neurodegeneration [10]. Påvisande av alzheimerpatologi görs med hjälp av biomarkörer, medan kognitiva symtom endast används för att gradera sjukdomens svårighetsgrad.
Likvoranalyser för alzheimer
Likvoranalyser av basala biomarkörer för alzheimer finns tillgängliga för klinisk diagnostik sedan flera år. Dessa biomarkörer följer A/T/N-klassificeringen och innefattar Aβ (Aβ42 och Aβ42/40-kvoten), fosforylerat tau (P-tau) och total-tau (T-tau), där kombinationen sänkt Aβ42 och Aβ42/40-kvot tillsammans med ökat T-tau och P-tau i likvor ofta kallas för den typiska »alzheimerprofilen« [11].
Patofysiologi och likvoranalyser
Aβ (Aβ42) är den dominerade formen i de amyloida plack som bildas i hjärnan vid alzheimer [6]. Sänkt Aβ42 i likvor korrelerar starkt med inbindningsgraden av amyloidligander mätt med PET-teknik [12], talande för att sänkt Aβ42 vid alzheimer orsakas av att peptiden fastnar i placken i hjärnvävnaden, vilket leder till lägre likvornivåer [13]. Samstämmigheten med kvantifiering av amyloid vid PET-undersökning (amyloid-PET) blir ännu bättre om man i stället använder Aβ42/40-kvoten [14-16], sannolikt på grund av att Aβ40, som är oförändrad vid alzheimer [17], normaliserar Aβ42-koncentrationen mellan personer med konstitutionellt låg eller hög total Aβ-produktion; en plackinducerad relativ minskning av Aβ42-koncentrationen kan därmed detekteras med större korrekthet [15].
Tau är ett neuronalt protein lokaliserat till axoner, och likvornivån av T-tau ses som en markör för intensiteten av neurodegeneration vid alzheimer, en tolkning som huvudsakligen baseras på hur nivån av T-tau förändras vid andra neurodegenerativa sjukdomar. Exempelvis ses en mycket uttalad ökning vid Creutzfeldt–Jakobs sjukdom, där det finns en mycket snabbt förlöpande neurodegeneration, men även att T-tau predicerar sjukdomens progresshastighet i olika faser av alzheimer. Likvornivån av P-tau är kopplad till fosforyleringsgraden av tau och därmed sannolikt även till utveckling av tau-patologi vid alzheimer. Till skillnad från T-tau förändras inte P-tau vare sig vid akut hjärnskada eller vid andra sjukdomar med taupatologi, och P-tau verkar därför vara en specifik markör för alzheimer. Både T- och P-tau-koncentrationerna är oftast normala vid andra tauopatier, såsom vissa former av frontallobsdemens och progressiv supranukleär paralys.
Ett mycket stort antal studier har konsekvent visat en hög diagnostisk träffsäkerhet av Aβ42, T-tau och P-tau i likvor för alzheimer [17]. Detta gäller även vid lindrig kognitiv svikt, där patienterna söker för minnesstörning eller i en del fall andra lindriga kognitiva symtom. Likvoranalyserna (lågt Aβ42 tillsammans med högt T-tau eller P-tau) kunde i en studie med 95 procents säkerhet predicera vilka patienter med lindrig kognitiv svikt som hade prodromal alzheimer [18], ett fynd som kort därefter kunde verifieras i flera stora studier [19-21]. Likvornivån av Aβ42 börjar sjunka redan innan de första symtomen visar sig [22], medan ökningen av T-tau och P-tau kommer något senare i sjukdomsförloppet i närmare anslutning till klinisk sjukdomsdebut [23]. Förutom en massiv klinisk validering har dessa likvoranalyser genomgått en teknisk utveckling, från tidiga manuella ELISA-metoder till dagens automatiserade instrument, vilket resulterat i en hög precision, också mellan olika kliniska laboratorier [24].
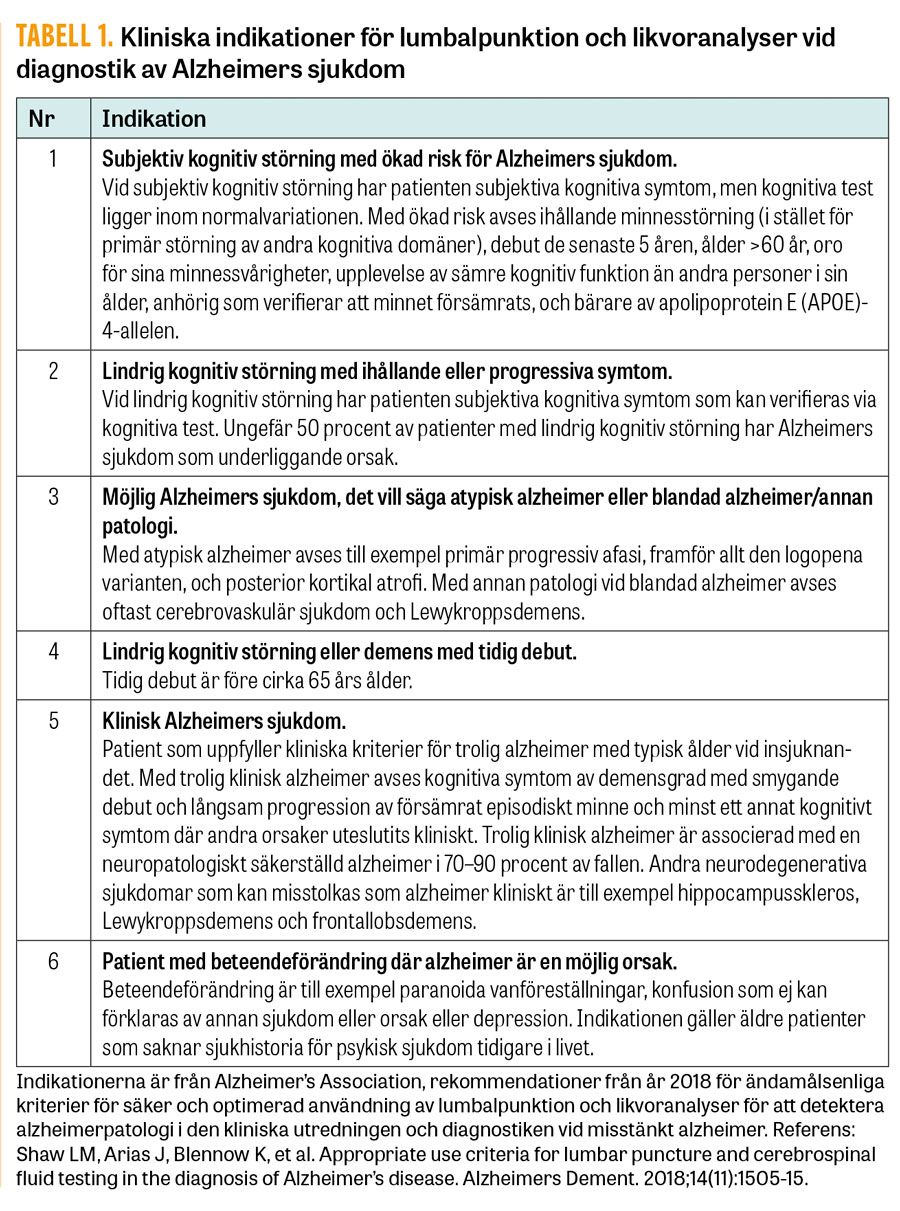
Nyligen har Alzheimer’s Association presenterat rekommendationer för användandet av likvoranalyser (Aβ42, T-tau och P-tau) i den kliniska utredningen och diagnostiken vid misstänkt Alzheimers sjukdom, se Tabell 1.
Synapsmarkörer för alzheimer
Synapsdegeneration är en viktig komponent i symtomutvecklingen vid alzheimer [25-27]. Synapsproteiner, som det presynaptiska SNAP-25 och det dendritiska proteinet neurogranin, utsöndras till likvor [28, 29]. Nivån av neurogranin i likvor är ökad vid alzheimer [30]. Högt neurogranin ses tidigt i sjukdomsförloppet och predicerar progress av kognitiva symtom [31]. Ett intressant och viktigt fynd är att ökat neurogranin verkar vara specifikt för alzheimer; det ses inte vid andra neurodegenerativa sjukdomar som pannlobsdemens eller Lewykroppsdemens [32, 33], vilket kan ha ett värde i differentialdiagnostiken.
Blodtest för alzheimer
Analystekniska framsteg har gjort det möjligt att mäta flera av alzheimermarkörerna i vanliga blodprov. Ett flertal artiklar har presenterat lovande resultat för koncentrationsbestämning av Aβ42 och Aβ42 i plasma med Simoa (Single molecule array)-teknik [34] samt immunprecipitering kombinerat med masspektrometri [35, 36], där en lägre Aβ42/40-kvot visade god samstämmighet med amyloid-PET. Även det axonala proteinet neurofilament light (NFL) kan mätas i blodprov med Simoa-teknik [37], och studier på alzheimer visar en ökad nivå av NFL i plasma samt en god överensstämmelse med NFL-koncentrationer i likvorprov [38]. I sammanhanget bör dock poängteras att NFL är en generell markör för neurodegeneration och annan nervcellsskada oavsett genes [39, 40]. En framtida applikation för plasma-NFL skulle kunna vara ett första screeningtest i den kliniska utvärderingen av patienter med misstänkt kognitiv sjukdom eller symtom på andra nervcellsskadande processer.
PET-kameratekniker för diagnostik
Under de senaste åren har det utvecklats reliabla PET-kameratekniker som kan detektera antingen Aβ- eller tau-aggregat i hjärnan hos levande människor. Den första liganden för Aβ-aggregat kallas för 11C-PiB (Pittsburgh compound B) [41] och har i ett stort antal forskningsstudier visats kunna påvisa Aβ-aggregat in vivo [42]. Då halveringstiden endast är 20 minuter har man utvecklat 18F-inmärkta ligander för Aβ-PET, vilka har en halveringstid på 110 min, varav tre stycken (Vizamyl, Amyvid och Neuraceq) nu är godkända för användning i klinisk rutinsjukvård. Alla dessa tre ligander har provats i studier av vård i livets slutskede, där man påvisat att de med över 90 procents säkerhet kan skilja personer drabbade av alzheimer från patienter med andra kognitiva sjukdomar [42]. Per definition har alla personer med alzheimer Aβ-patologi, och undersökningen har en mycket hög sensitivitet och därmed högt negativt prediktivt värde, det vill säga att man med hög säkerhet kan utesluta alzheimer om Aβ-PET är normal, oavsett ålder och symtombild hos patienten.
Vi och andra forskargrupper har emellertid visat att Aβ-patologi uppstår i hjärnan 15–20 år innan en person med alzheimer utvecklar demens. Med tanke på att cirka 20–25 procent av personer som är 90–100 år har demens sekundär till alzheimer så är det inte oväntat att 20–25 procent av personer som är cirka 80 år har en patologisk Aβ-PET även om de inte har några kognitiva symtom. Mot denna bakgrund har metoden en lägre specificitet och därmed lägre positivt prediktivt värde för alzheimer hos patienter med kognitiva symtom som är äldre, det vill säga cirka 80 år och uppåt, och därför ger en patologisk Aβ-PET hos äldre personer (>80 år) en något osäker information.
Ett flertal studier har undersökt samstämmigheten mellan likvoranalyser och amyloid-PET och funnit en mycket hög konkordans (runt 90 procent) mellan likvor-Aβ42 och amyloid-PET [43], inkluderande stora prospektiva studier på konsekutiva patienter som remitterats till en minnesklinik för utredning [44]. Valet mellan likvortest och Aβ-PET kan därför göras baserat på faktorer som tillgänglighet, kostnader, riskskattning (postpunktionshuvudvärk vs exponering för strålning) samt läkarens och patientens egen preferens.
Till skillnad från Aβ-patologi, som vid alzheimer startat 15–20 år före demens, så är utbredd kortikal tau-patologi starkt associerad med nervcellsdöd, och denna process sker i nära samband med att de drabbade patienterna utvecklar tydliga kognitiva symtom [45]. Ett flertal 18F-ligander har nu utvecklats som kan detektera alzheimerrelaterad tau-patologi. Tau-PET-mätning kan med mycket stor säkerhet påvisa om en person med demens har alzheimer eller inte, det vill säga specificiteten för alzheimer är högre än för Aβ-PET eller likvorbaserade markörer under demensfasen av sjukdomen, och detta oavsett ålder [46]. Preliminära data tyder även på att tau-PET utförd i livet korrelerar väl med tau-patologi detekterat med immunhistokemi efter döden [47]. Dock är det ännu oklart hur bra tau-PET är på att detektera alzheimer under de tidiga faserna av sjukdomen innan demens har utvecklats, och för att bestämma detta krävs longitudinella studier, vilka är på gång. Initiala resultat tyder dock på att Aβ-PET och likvoranalys av Aβ42/Aβ40 alternativt P-tau/Aβ42 har högre diagnostisk säkerhet under de tidiga faserna av alzheimer. Ännu är ingen tau-PET-ligand godkänd av regulatoriska myndigheter i USA eller Europa.
Behandling
Kognitionsförbättrande medel
Många typer av neurotransmittor-abnormaliteter uppstår, vilka påverkar kolinerga, monoaminerga och glutamaterga system [48]. Två klasser av kognitionsförstärkande läkemedel har godkänts för användning vid alzheimer: kolinesterashämmare (AChEI) och N-metyl-D-aspartat (NMDA)-receptorantagonist (memantin). AChEI minskar den extrasynaptiska metabolismen av acetylkolin, ökar uppehållstiden för neurotransmittorn och förbättrar postsynaptisk stimulering. AChEI inkluderar donepezil, rivastigmin och galantamin. Donepezil och galantamin hämmar enbart acetylkolinesteras, medan rivastigmin hämmar både acetyl- och butyrylkolinesteras. AChEI kombineras ofta med memantin, som verkar på det glutamaterga systemet genom att hämma NMDA-receptorn och därmed normalisera neurotransmittor-abnormaliteter [49]. Denna kombinationsterapi ger additiva fördelar [50].
En majoritet av patienterna får förbättrad kognitiv förmåga efter påbörjad terapi [51, 52]. Långtidsstudier tyder på fortsatt nytta av terapi trots kognitiv försämring över tid, med mindre försämring observerad hos patienter som fått behandling jämfört med dem som inte fått behandling. Nyligen har en metaanalys föreslagit individualisering av AChEI-terapi i relation till kön, apolipoprotein E och ålder [53]. Utsättning av terapi bestäms vanligtvis när patienten har nått en nivå av försämring där kognitionsförstärkande terapi inte längre ger någon positiv inverkan på livskvaliteten.
En restriktiv hållning bör gälla vad beträffar läkemedel med antikolinerg verkan, såsom urinvägsspasmolytika, äldre antihistaminer, neuroleptika, vissa antiparkinsonmedel och tricykliska antidepressiva.
Utveckling av immunterapi riktad mot Aβ och tau
Immunterapi mot Aβ.I slutet av 1990-talet inledde utvecklingen av immunterapi mot Alzheimers sjukdom. På transgena möss lyckades forskare visa att Aβ-patologi kunde motverkas genom att vaccinera med Aβ. Den första studien illustrerade både en preventiv effekt och att man kunde få redan befintliga plack att tillbakabildas [54]. Denna upptäckt resulterade i en fas 1-prövning, som byggde på injektion av Aβ-fibriller intramuskulärt på patienter, men studien fick avbrytas efter att ungefär 5 procent av de deltagande patienterna utvecklat inflammatoriska hjärnförändringar. De patienter som uppvisat en hög antikroppstiter hade försämrats långsammare än övriga patienter [55]. Därtill visade neuropatologiska analyser en tydligt minskad amyloidinlagring i hjärnan jämfört med individer som erhållit placebo [56].
I syfte att minska biverkningsrisken påbörjades i stället försök baserade på passiv immunterapi genom tillförsel av monoklonala antikroppar mot Aβ. Efter att ett flertal lyckade studier genomförts på musmodeller kunde de första kliniska prövningarna inledas. Det visade sig snart att även denna form av behandling var behäftad med biverkningar, framför allt perivaskulära ödem och mikroblödningar, som tillsammans kallas »amyloid related imaging abnormalities« (ARIA).
Kartläggningen av mutationer i genen för APP har bidragit till viktig kunskap om hur sjukdomen utvecklas. Studier av den »svenska« mutationen visade för första gången i en klinisk situation att Aβ initierar sjukdomsprocessen [57]. I slutet av 1990-talet hittades en ytterligare mutation i APP hos patienter i Norrland, som döptes till den »arktiska« mutationen. Det visade sig att lösliga, aggregerade former av Aβ ökade genom denna mutation och att dessa så kallade protofibriller var särskilt neurotoxiska [58] (Figur 1).
En protofibrill är uppbyggd av endast cirka 20–150 enskilda Aβ-molekyler, och dessa strukturer, som kan liknas vid »ostbågar«, går att visualisera med högupplösande atomkraftsmikroskopi (Figur 2). Protofibriller bildas också hos övriga alzheimerpatienter, men i mindre utsträckning än hos individer med den arktiska mutationen [59]. Baserat på fynden kring den arktiska mutationen utvecklades vid Uppsala universitet en konformationsberoende monoklonal antikropp, mAb158, som känner igen en unik struktur i Aβ-protofibrillen [60]. Studier på transgena möss utfördes och visade goda behandlingseffekter med denna antikropp [61, 62].
Kliniska studier med humaniserad antikropp BAN2401 inleddes 2010 och kunde genomföras utan några allvarliga biverkningar [63]. En stor fas 2-studie med alzheimerpatienter i tidiga stadier avslutades i juli 2018 efter att 856 patienter hade deltagit i 18 månader [64]. En dosberoende minskning av Aβ-plack i hjärnan kunde ses efter 18 månader, och av patienter som fick den högsta dosen blev 93 procent amyloid-PET-negativa. Även effekter i likvor kunde påvisas. Värden av P-tau, neurogranin och NFL påverkades alla på ett gynnsamt sätt. Vidare kunde man, efter 18 månaders behandling, påvisa 26–47 procents minskning i försämringstakten [64]. En fas 3-studie med BAN2401 inleddes under våren 2019 och beräknas vara avslutad år 2022.
Ytterligare två antikroppar för behandling av alzheimer befinner sig i fas 3-prövningar. Bioteknikföretaget Biogen har nyligen rapporterat positiva effekter med sin antikropp, aducanumab, och avser att ansöka om registrering av detta preparat i USA under 2020. Även företaget Roches antikropp gantenerumab har haft en besvärlig väg framåt. En tidigare fas 3-studie misslyckades, men nu pågår en ny studie med en högre dos av antikroppen.
Immunterapi mot tau. Det pågår även en utveckling av immunterapi riktad mot tau. Denna strategi kan motiveras utifrån observationen att tau-patologin i hjärnan hos personer med alzheimer korrelerar bättre än plackpatologin med den kliniska bilden.
Studier på transgena möss med antingen aktiv eller passiv immunterapi mot tau har både kunnat minska hjärnpatologin och dämpa symtomen [65-68]. Vid den aktiva immunterapin har man huvudsakligen nyttjat oligomera eller fibrillära former av tau, och för passiv immunterapi har man använt antikroppar mot olika epitoper och konformationer av tau-molekylen.
En första klinisk prövning med ett tau-vaccin kunde initieras 2013. Flertalet studier pågår fortfarande [69], men hittills har ingen av dessa kunnat rapportera gynnsamma effekter.
Icke-farmakologiska interventioner
Nyligen har en banbrytande finsk-svensk klinisk prövning visat att en intervention riktad mot kardiovaskulära riskfaktorer tillsammans med dietoptimering samt både fysisk och kognitiv träning kan förbättra kognitiv funktion hos friska äldre [70]. Andra icke-farmakologiska interventioner som testats inkluderar minnesträning, mental och social stimulering, fysiska träningsprogram samt vårdgivarutbildning. Dessa kan möjligen förbättra människors kognitiva prestanda och öka deras oberoende. Det är viktigt att se till att sådana interventioner inte kräver för mycket av personen och inte är stressande. Exempel på kognitiv träning för patienter med lindrig till måttlig alzheimer inkluderar aritmetiska problem och övningar där nummerserier ska slutföras, eller memorering och igenkänning av bilder. Dessa övningar erbjuds antingen en-mot-en eller i grupp, vanligtvis 1–2 gånger per vecka i 30–90 minuter [71].
Flera andra icke-farmakologiska interventioner har föreslagits, såsom verklighetsorienteringsträning, där patienten ges grundläggande uppgifter upprepade gånger, till exempel att minnas namn, datum eller tid, med syfte att förbättra orienteringen i tid och rum. Eventuella negativa effekter av sådan träning måste övervägas; exempelvis kan det vara mycket frustrerande för patienter med demens att upprepade gånger inte lyckas genomföra en övning [72]. Sociala aktiviteter kan förbättra livskvaliteten för patienter och deras familjemedlemmar och kan tänkas minska apati och därmed behovet av vård [73].
Fysisk aktivitet såsom promenader, styrketräning och uthållighetsövningar med cirka 2–3 träningspass per vecka i 30–60 minuter kan leda till att patienter med demens kan bibehålla sin rörlighet under en längre tid [74].
Utbildningsprogram för vårdgivare har utvecklats för att ge icke-professionella vårdgivare stöd. Syftet med dessa program är att lära ut hur man kan hjälpa människor med alzheimer att behålla sina förmågor så länge som möjligt, så att de bättre kan hantera situationer som är stressande och upprörande [75].
Medelhavsdiet har föreslagits ha en positiv effekt på minne och kognitiv förmåga, men det finns inget vetenskapligt bevis för att medelhavsdiet kan förhindra eller bromsa alzheimer [76]. Kosttillskott påstås förbättra mentala prestanda, men varken fiskoljekapslar (omega-3-fettsyror) eller andra kosttillskott har visats ha någon effekt vid alzheimer [77].
Potentiella bindningar eller jävsförhållanden: Lars Lannfelt och Hans Basun är anställda av Bioarctic. Lars Lannfelt är grundare och sitter i styrelsen. Martin Ingelsson sitter i vetenskapliga rådet för Bioarctic. Henrik Zetterberg har varit konsult åt Samumed, Wave, Cog Rx och Roche Diagnostics, har föreläst på seminarier som sponsrats av Alzecure och Biogen samt är en av grundarna av Brain Biomarker Solutions in Gothenburg AB, ett GU Ventures-bolag vid Göteborgs universitet. Kaj Blennow har varit konsult åt Abcam, Axon, Biogen, Lilly, Mag Qu, Novartis och Roche Diagnostics samt är en av grundarna av Brain Biomarker Solutions in Gothenburg AB. Nenad Bogdanovic har varit senior medical director på Wyeth och Pfizer samt är ordförande för de externa säkerhetskommittéerna vid Orion Pharma och Kyowa-Kirin.
(uppdaterad 2020-03-16)
Referenser
- Nationella riktlinjer för vård och omsorg vid demenssjukdom. Stockholm: Socialstyrelsen; 2017. Artikelnr 2017-12-2.
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet 2006:368(9533):387-403.
- Bogdanovic N. Alzheimer’s disease: towards a multifaceted approach in neuropathological diagnosis [avhandling]. Stockholm: Karolinska institutet; 1998.
- Hardy J, Selkoe D. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353-6.
- Fratiglioni L, Qiu CX. Epidemilogy of dementia. In: Dening T, Thomas A (editors). Oxford textbook of old age psychiatry. 2nd ed. Oxford: Oxford University Press; 2013. p. 389-413.
- Masters CL, Bateman R, Blennow K, et al. Alzheimer’s Disease. Nat Rev Dis Primers. 2015;1:15059.
- Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13(6):614-29.
- Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960-9.
- Kovacs GG, Milenkovic I, Wöhrer A, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol. 2013;126(3):365-84.
- Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535-62.
- Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med. 2018;284(6):643-63.
- Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59(3):512-9.
- Andreasen N, Hesse C, Davidsson P, et al. Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56(6):673-80.
- Janelidze S, Zetterberg H, Mattsson N, et al; Swedish BioFINDER study group. CSF Abeta42/Abeta40 and Abeta42/Abeta38 ratios: better diagnostic markers of Alzheimer disease. Ann Clin Transl Neurol. 2016;3(3):154-65.
- Lewczuk P, Matzen A, Blennow K, et al. Cerebrospinal fluid Aβ42/40 corresponds better than Aβ42 to amyloid PET in Alzheimer’s disease. J Alzheimers Dis. 2017;55(2):813-22.
- Pannee J, Gobom J, Shaw LM, et al. Round robin test on quantification of amyloid-beta 1-42 in cerebrospinal fluid by mass spectrometry. Alzheimers Dement. 2016;12(1):55-9.
- Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673-84.
- Hansson O, Zetterberg H, Buchhave P, et al. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5(3):228-34.
- Visser PJ, Verhey F, Knol DL, et al. Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol. 2009;8(7):619-27.
- Shaw LM, Vanderstichele H, Knapik-Czajka M, et al; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403-13.
- Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302(4):385-93.
- Skoog I, Davidsson P, Aevarsson O, et al. Cerebrospinal fluid beta-amyloid 42 is reduced before the onset of sporadic dementia: a population-based study in 85-year-olds. Dement Geriatr Cogn Disord. 2003;15(3):169-76.
- Stomrud E, Hansson O, Blennow K, et al. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord. 2007;24(2):118-24.
- Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β-amyloid (1-42) in human cerebrospinal fluid. Alzheimers Dement. 2016;12(5):517-26.
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457-64.
- Blennow K, Bogdanovic N, Alafuzoff I, et al. Synaptic pathology in Alzheimer’s disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J Neural Transm (Vienna). 1996;103(5):603-18.
- Sze CI, Troncoso JC, Kawas C, et al. J. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56(8):933-44.
- Davidsson P, Jahn R, Bergquist J, et al. Synaptotagmin, a synaptic vesicle protein, is present in human cerebrospinal fluid: a new biochemical marker for synaptic pathology in Alzheimer disease? Mol Chem Neuropathol. 1996;27(2):195-210.
- Davidsson P, Puchades M, Blennow K. Identification of synaptic vesicle, pre- and postsynaptic proteins in human cerebrospinal fluid using liquid-phase isoelectric focusing. Electrophoresis. 1999;20(3):431-7.
- Thorsell A, Bjerke M, Gobom J, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010;1362:13-22.
- Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11(10):1180-90.
- Portelius E, Olsson B, Höglund K, et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018;136(3):363-76.
- Wellington H, Paterson RW, Portelius E, et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016;86(9):829-35.
- Janelidze S, Stomrud E, Palmqvist S, et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci Rep. 2016;6:26801.
- Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018;554(7691):249-54.
- Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13(8):841-9
- Gisslén M, Price RW, Andreasson U, et al. Plasma concentration of the neurofilament light protein (NFL) is a biomarker of CNS injury in HIV infection: a cross-sectional study. EBioMedicine. 2016;3:135-40.
- Mattsson N, Andreasson U, Zetterberg H, et al; Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s disease neuroimaging I. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74(5):557-66.
- Rohrer JD, Woollacott IO, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87(13):1329-36.
- Rojas JC, Karydas A, Bang J, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. 2016;3(3):216-25.
- Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306-19.
- Perani D, Iaccarino L, Lammertsma AA, et al. A new perspective for advanced positron emission tomography-based molecular imaging in neurodegenerative proteinopathies. Alzheimers Dement. 2019;15(8):1081-103.
- Janelidze S, Zetterberg H, Mattsson N, et al; Swedish BioFINDER study group. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: better diagnostic markers of Alzheimer disease. Ann Clin Transl Neurol. 2016;3(3):154-65.
- Rabinovici GD, Gatsonis C, Apgar C, et al. Association of amyloid positron emission tomography with subsequent change in clinical management among medicare beneficiaries with mild cognitive impairment or dementia. JAMA. 2019;321(13):1286-94.
- Savva GM, Wharton SB, Ince PG, et al; Medical Research Council Cognitive Function and Ageing Study. Age, neuropathology, and dementia. N Engl J Med. 2009;360(22):2302-9.
- Ossenkoppele R, Rabinovici GD, Smith R, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2018;320(11):1151-62.
- Smith R, Wibom M, Pawlik D, et al. Correlation of in vivo [18F]flortaucipir with postmortem alzheimer disease tau pathology. JAMA Neurol. 2019;76(3):310-37.
- Noetzli M, Eap CB. Pharmacodynamic, pharmacokinetic and pharmacogenetic aspects of drugs used in the treatment of Alzheimer’s disease. Clin Pharmacokinet. 2013;52(4):225-41.
- Parsons CG, Danysz W, Dekundy A, et al. Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox Res. 2013;24(3):358-69.
- Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-24.
- Takeda A, Loveman E, Clegg A, et al. A systematic review of the clinical effectiveness of donepezil, rivastigmine and galantamine on cognition, quality of life and adverse events in Alzheimer’s disease. Int J Geriatr Psychiatry. 2006;21(1):17-28.
- Di Santo SG, Prinelli F, Adorni F, et al. A meta-analysis of the efficacy of donepezil, rivastigmine, galantamine, and memantine in relation to severity of Alzheimer’s disease. J Alzheimers Dis. 2013;35(2):349-61.
- Lane RM, Darreh-Shori T. Understanding the beneficial and detrimental effects of donepezil and rivastigmine to improve their therapeutic value. J Alzheimer Dis. 2015;44(4):1039-62.
- Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173-7.
- Hock C, Konietzko U, Streffer JR, et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38(4):547-54.
- Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64(9):1563-72.
- Mullan M, Crawford F, Axelman K, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1(5):345-7.
- Nilsberth C, Westlind-Danielsson A, Eckman CB, et al. The »Arctic« (E693G) mutation causes Alzheimer’s disease by increasing Aβ protofibril formation. Nat Neurosci. 2001;4(9):887-93.
- Johansson AS, Berglind-Dehlin F, Karlsson G, et al. Physiochemical characterization of the Alzheimer’s disease-related peptides A beta 1-42Arctic and A beta 1-42wt. FEBS J. 2006;273(12):2618-30.
- Englund H, Sehlin D, Johansson AS, et al. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. 2007;103(1):334-45.
- Lord A, Gumucio A, Englund H, et al. An amyloid-beta protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2009;36(3):425-34.
- Tucker S, Möller C, Tegerstedt K, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis. 2015;43(2):575-88.
- Logovinsky V, Satlin A, Lai R, et al. Safety and tolerability of BAN2401 – a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8(1):14.
- Swanson CJ, Zhang Y, Dhadda S, et al. Treatment of early AD subjects with BAN2401, an anti-Aβ protofibril monoclonal antibody, significantly clears amyloid plaque and significantly reduces clinical decline [abstract DT-01-07]. Alzheimer’s Association International Conference (AAIC), Chicago, 22–26 jul 2018.
- Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30(49):16559-66.
- Castillo-Carranza DL, Gerson JE, Sengupta U, et al. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014;40(Suppl 1):S97-111.
- Liu W, Zhao L, Blackman B, et al. Vectored intracerebral immunization with the anti-tau monoclonal antibody PHF1 markedly reduces tau pathology in mutant tau transgenic mice. J Neurosci. 2016;36(49):12425-35.
- Albert M, Mairet-Coello G, Danis C, et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain. 2019;142(6):1736-50.
- Hoskin JL, Sabbagh MN, Al-Hasan Y, et al. Tau immunotherapies for Alzheimer’s disease. Exp Opin Investig Drugs. 2019;28(6):545-54.
- Ngandu A, Lehtisalo L, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015; 385(9984):2255-63.
- Hill NT, Mowszowski L, Naismith SL, et al. Computerized cognitive training in older adults with mild cognitive impairment or dementia: a systematic review and meta-analysis. Am J Psychiatry. 2017;174(4):329-40.
- Camargo C, Justus F, Retzlaff G. The effectiveness of reality orientation in the treatment of Alzheimer’s disease. Neurology. 2015;84(14 Suppl):181-6. Am J Alzheimers Dis Other Demen. 2015;30(5):527-32.
- Leung P, Orrell M, Orgeta V. Social support group interventions in people with dementia and mild cognitive impairment: a systematic review of the literature. Int J Geriatr Psychiatry. 2015;30(1):1-9.
- Forbes D, Forbes SC, Blake CM, et al. Exercise programs for people with dementia. Cochrane Database Syst Rev. 2015;(4):CD006489.
- Klimova B, Valis M, Kuca K, et al. E-learning as valuable caregivers’ support for people with dementia – a systematic review. BMC Health Serv Res. 2019;19(1):781-8.
- Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
- Burckhardt M, Herke M, Wustmann T, et al. Omega-3 fatty acids for the treatment of dementia. Cochrane Database Syst Rev. 2016;(4):CD009002.
Engelsk sammanfattning
Alzheimer’s disease is the most common cause of dementia. As many as 250,000 people in Sweden will have a dementia disease in 2050. The »amyloid cascade hypothesis« is a common model which explains how β-amyloid affects the function of the nerve cells. Alzheimer’s disease has a long-lasting course and can present in typical and atypical forms. CSF analyses for »core AD CSF biomarkers« and synaptic proteins have been available for clinical diagnostics. PET scanning can detect either β-amyloid or tau aggregates in the brain of living humans. Current Alzheimer’s disease therapy is based on two classes of cognition-enhancing drugs: acetylcholinesterase inhibitor and NMDA-receptor antagonist, which delays cognitive decline in most patients. The latest clinical development of potential therapy for Alzheimer’s is active or passive immunotherapy against brain β-amyloid and tau, where several studies have shown varying but promising treatment effects. Non-pharmacological interventions in patients with AD aim to delay the loss of mental abilities, helping people to be independent in everyday life for as long as possible, and to increase their well-being and quality of life.