
Huntingtons sjukdom är en autosomalt dominant neurodegenerativ sjukdom, som leder till förtidig död. Orsaken är expansion av en CAG-triplett i huntingtingenen.
Sjukdomen är vanligast i västerländska befolkningar. Den debuterar typiskt i medelåldern och orsakar framskridande motoriska, kognitiva och psykiska symtom.
I dag finns endast symtomlindrande mediciner tillgängliga, men ny molekylärgenetisk teknologi kan komma att möjliggöra behandlingar som minskar nivåerna av muterat huntingtin.
Huntingtons sjukdom är en autosomalt dominant nedärvd neurodegenerativ sjukdom. Sjukdomen består av en triad av motoriska, psykiska och kognitiva symtom. De motoriska symtomen består tidigt i förloppet ofta av framträdande ofrivilliga rörelser (korea), medan bradykinesi och dystoni tenderar att prägla senare sjukdomsfas då även gångförmågan upphör. Dysartri och dysfagi är vanligt. De psykiska symtomen är varierande och innefattar depression, ångest och psykotiska symtom. Den kognitiva dysfunktionen är av exekutiv karaktär med försämrad organisationsförmåga, koncentration och uppmärksamhet samt bristande initiativförmåga (apati) [1].
Det är nu snart 150 år sedan George Huntington karakteriserade sjukdomen och dess ärftlighetsmönster väl i sin essä om sjukdomen (»On chorea«, 1872). Detta var innan Gregor Mendels publikation om »ärftlighetslagar« hade fått spridning och långt innan begrepp som »autosomalt dominant« myntats. Den sjukdomsorsakande genmutationen på kromosom 4 kartlades år 1993 – en trinukleotidexpansion av CAG (som kodar för aminosyran glutamin) i huntingtingenen [2].
Huntingtinproteinets funktion är komplex och ofullständigt klarlagd. Det står dock klart att proteinet är avgörande för nervsystemets utveckling och inblandat i en rad interaktioner med andra proteiner. När antalet CAG-repetitioner är 40 eller högre är penetransen fullständig, och debutåldern korrelerar negativt med repetitionslängd (dvs tidigare debutålder är associerad med högre repetitionslängd). Alleler mellan 36 och 39 repetitioner går med nedsatt penetrans och medför risk för sjukdom först vid hög ålder. Sjukdomen kan debutera redan i tonåren, men vanligtvis i 35–55 års ålder.
Huntingtons sjukdom är den vanligaste autosomalt dominant nedärvda formen av demens i den västerländska världen. Prevalensen är högst i befolkningar av europeiskt ursprung, och sjukdomen förekommer med betydligt lägre prevalens i andra regioner i världen. Nationella prevalenssiffror i Sverige saknas, men förekomsten av sjukdomen har i modernare studier uppskattats till 10–15 per 100 000 personer i anglosaxiska länder [3, 4].
Normalt finns upp till 26 repetitioner på vardera huntingtinallel. Sjukdomens prevalens är associerad med medelvärdet av CAG-repetitioner i bakgrundsbefolkningen. Intermediäralleler (27–35 repetitioner) är relativt vanliga i befolkningen (6–7 procent) och orsakar inte Huntingtons sjukdom, men ger möjligen risk för beteendestörning och/eller depression. Intermediäralleler är instabila, varför ytterligare expansion under meios (typiskt via fädernet) kan resultera i sjukdomsorsakande expansion i nästa generation. Således förekommer fall utan påvisbar sjukdom hos föräldrar, dvs nymutationer [5]. Samma fenomen förklarar en trend mot tidigare sjukdomsdebut i släkter med Huntingtons sjukdom (antecipation).
Neuropatologi
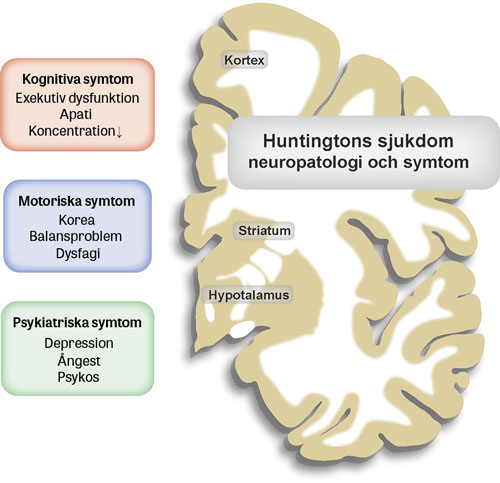
Huntingtin finns i alla kroppens vävnader, men det är ändå i striatum i de basala ganglierna i hjärnan som den mest uttalade skadan sker med nervcellsdöd och atrofi av vävnaden. Parallellt sker dessutom degeneration av både vitsubstans och hjärnbark. De motoriska symtomen är kopplade till neurodegeneration i striatum, medan den kognitiva dysfunktionen är associerad till patologi i hjärnbarken.
De neurobiologiska förändringarna som ligger bakom de psykiska symtomen är inte klarlagda men kan bestå av patologi i hjärnbarken och hypotalamus med förlust av känsloreglerande ämnen som oxytocin och orexin (hypokretin) [6] (Figur 1). En rad cellulära mekanismer påverkas av expansionen på huntingtingenen [7]. Sannolikt drivs majoriteten av skadeverkan av ökad och giftig funktion på grund av expansionen, men även bortfall av normal huntingtinfunktion verkar spela en viss roll.
Själva polyglutaminfragmentet klyvs av från huntingtinproteinet och transporteras in i cellkärnan, där det stör den normala gentranskriptionen. Huntingtinproteinet ansamlas i intracellulära inklusioner på samma sätt som man ser andra protein göra vid ett flertal neurodegenerativa sjukdomar [8]. Axonal- och vesikeltransport påverkas, vilket bl a minskar nivåerna av tillväxtfaktorn BDNF (hjärnrelaterad neurotrofisk faktor; brain derived neurotrophic factor) i striatum. Störda processer i synaptisk transmission, excitotoxicitet (toxisk effekt av för höga glutamatnivåer) liksom påverkan på mitokondrier kan också vara delar av sjukdomsmekanismer.
Forskning talar för att reparationsmekanismerna av DNA är påverkade, vilket leder till att mängden CAG kan öka upp till flera hundra i drabbade nervceller i hjärnan vid Huntingtons sjukdom, som en följd av s k somatisk instabilitet. Stora genetiska studier har också visat att förändringar i gener som tillverkar DNA-reparationsfaktorer bidrar till att avgöra insjuknandeålder [9]. Detta är ännu ett spännande spår i forskningen om Huntingtons sjukdom, där man försöker hitta nya potentiella angreppspunkter för välbehövda sjukdomsbromsande behandlingar.
Sjukdomsprogression och biomarkörer
Biomarkörer eftersöks för att objektivt kunna utvärdera effekten av potentiella behandlingar. Graden av motoriska och kognitiva symtom kan skattas med olika instrument, vilket i dag är utfallsmåttet i kliniska prövningar. Skalorna har dock fallgropar såsom variation mellan olika undersökare och patientens dagsform.
Robusta biomarkörer som kan mäta sjukdomsprogression skulle underlätta utvecklingen av sjukdomsmodifierande behandlingar. Av olika MR-parametrar har progressiv atrofi av striatum visat sig vara en robust surrogatmarkör för sjukdomsprogression, som kan uppmätas 15 år före sjukdomsdebut. Mätmetoder för koncentration av muterat huntingtinprotein i likvor har utvecklats, vilket är användbart i läkemedelsstudier där målet är just minskning av dessa nivåer.
Nervcellsskademarkören neurofilament light (NFL), som även implementerats i kliniken vid andra neurologiska sjukdomar, har visat sig kunna förutsäga svårighetsgraden av symtomen vid Huntingtons sjukdom [10].
Omhändertagande av patienter med Huntingtons
Rådande konsensus i Sverige är att patienter med Huntingtons sjukdom ska få tillgång till sjukvårdsteam med spetskompetens om sjukdomen. Huntingtonteam finns etablerade i Göteborg, Lund, Stockholm, Umeå och Uppsala. Eftersom sjukdomens symtom är mångskiftande behövs neurolog, psykiater, psykolog, sjuksköterska, fysioterapeut, arbetsterapeut, logoped, dietist och kurator i dessa team [11].
Patientens dominerande symtombild, som varierar individer emellan, kartläggs bäst av teamets olika professioner och tas upp vid teamkonferens. Problem identifieras och relevanta åtgärder vidtas. Patienter som får diagnosen bör erbjudas remiss för bedömning och uppföljning hos ett av landets Huntingtonteam.
Sjukdomens progredierande natur och svåra prognos fordrar regelbunden uppföljning eftersom symtomen förändras över tid. Sjukdomen medför gradvis förlust av förmågor och förkortad överlevnad. Regelbunden uppföljning behövs, och i perioder kan symtomen vålla stora besvär som kräver extra täta kontakter och besök. Detta i kombination med Sveriges geografi tillåter inte en fullständig centralisering till enstaka centrum eftersom avstånden blir för långa för patienterna. I sjukdomens senare fas är det inte sällan svårt för patienter att resa till de högspecialiserade teamen, och vården får då organiseras med närmast belägna neurologklinik och/eller primärvård i samråd med Huntingtonteamet i fråga.
Evidensen för symtomlindrande farmaka är framför allt baserad på mindre studier och klinisk erfarenhet. Vanliga läkemedel mot depression och ångest är selektiva serotoninåterupptagshämmare (SSRI) och mirtazapin, som även kan lindra insomni. Mot besvärande korea används ofta andra generationens antipsykotika (olanzapin och risperidon) eller det för hyperkinetiska rörelserubbningar godkända läkemedlet tetrabenazin (hämmare av den vesikulära monoamintransportören). Nyttan av speciell antikoreatisk behandling bör vägas mot risken för betydande biverkningar.
Prediktiv gentestning
Prediktiv gentestning finns tillgänglig vid universitetsklinikerna i landet. Den som genom ärftlighet har risk att bära på anlaget bör erbjudas remiss för att få information om ärftlighet och möjlighet till genetiskt test, vilket förutsätter ålder över 18 år. Väletablerade riktlinjer för hur testning bör gå till finns och bör följas [12]. Tyvärr sker ibland testning utan genetisk vägledning hos vårdgivare som inte har kännedom om testproceduren och dess fallgropar. Studier visar att endast omkring 15 procent av dem som löper 50 procents risk att insjukna genomgår prediktiv gentestning [13].
Motivationen till att genomgå testning är högst personlig, och komplexa situationer kan uppstå i samband med testning. I samband med graviditet efterfrågas ibland fostervattenprov, andra gånger planeras preimplantatorisk genetisk diagnostik (PGD), vilken utförs på Karolinska universitetssjukhuset i Huddinge och Sahlgrenska universitetssjukhuset i Göteborg. Om man inte vill avslöja den blivande förälderns genetiska status erbjuds exklusionstest på fostret.
Potentiella sjukdomsmodifierande behandlingar
Det är över 25 år sedan huntingtingenen isolerades, men efter drygt 100 kliniska prövningar finns fortfarande ingen sjukdomsmodifierande behandling. Dock finns nu anledning till optimism. Flertalet tidigare läkemedelsprövningar testade läkemedel som inte var specialdesignade för Huntingtons sjukdom. I dag pågår ett antal läkemedelsprövningar, och en del inriktas mot att sänka huntingtinnivåerna (Figur 2) (för översikt över pågående studier se [14]).
I en fas 1-/2a-studie av antisensoligonukleotiden (ASO) IONIS-HTTRx sågs en dosberoende minskning av muterat huntingtin i likvor hos patienter med Huntingtons sjukdom. Läkemedlet binder till huntingtin-mRNA och orsakar degradering. Det primära utfallsmåttet, säkerhet och tolerans uppnåddes [15].
En fas 3-studie har nu påbörjats och siktar på att inkludera mer än 600 patienter. Väl att märka hämmar preparatet (som nu betecknas RG6042) produktionen av både muterat huntingtin och normallängdsallelen. En potentiell fördel vore att i stället selektivt hämma muterat huntingtin och således undvika förlust av eventuellt viktig funktion av det normala proteinet. Prövningar med selektiv hämning inkluderade patienter redan under 2019. Dessa ASO-preparat ges intratekalt via lumbalpunktion vid regelbundna tidpunkter (längsta möjliga intervall utvärderas i studierna; möjligen blir det kvartalsvis).
Genterapi för att dämpa uttrycket av huntingtingenen i nervceller i striatum påbörjades under 2019 i en prövning av företaget Uniqure. Genterapi kan innebära behandling av en kronisk sjukdom i en enda seans. Optimalt vore dock om behandlingen var icke-invasiv. Dessutom finns risken att eventuella negativa effekter av terapin inte går att reversera.
Om något av ASO-preparaten visar effekt och når marknaden kommer det inte bara innebära en stor läkemedelskostnad, utan även fordra att intratekal behandling rent praktiskt kan erbjudas patienterna i landet med regelbundenhet. Om verksam behandling finns att tillgå sänks rimligen tröskeln för dem som överväger att genomgå prediktiv gentestning, varför man kan förvänta att fler personer får diagnosen tidigare. Detta skulle sammantaget innebära en omställning av huntingtonvården likt genombrottet för multipel skleros.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
(uppdaterad 2020-03-16)
Referenser
- Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204-16.
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971-83.
- Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord. 2014;29(1):105-14.
- Evans SJ, Douglas I, Rawlins MD, et al. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry. 2013;84(10):1156-60.
- Semaka A, Kay C, Doty C, et al. CAG size–specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet. 2013;50(10):696-703.
- Gabery S, Murphy K, Schultz K, et al. Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropa-thological analyses. Acta Neuropathol. 2010;120(6):777-88.
- Caron NS, Dorsey ER, Hayden MR. Therapeutic approaches to Huntington disease: from the bench to the clinic. Nat Rev Drug Discov. 2018;17(10):729-50.
- DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990-3.
- Jones L, Houlden H, Tabrizi SJ. DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 2017;16(1):88-96.
- Niemela V, Landtblom AM, Blennow K, et al. Tau or neurofilament light – which is the more suitable biomarker for Huntington’s disease? PLoS One. 2017;12(2):e0172762.
- Novak MJ, Tabrizi SJ. Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297-323.
- MacLeod R, Tibben A, Frontali M, et al; Editorial Committee and Working Group »Genetic Testing Counselling« of the European Huntington Disease Network. Recommendations for the predictive genetic test in Huntington’s disease. Clin Genet. 2013;83(3):221-31.
- Tassicker RJ, Teltscher B, Trembath MK, et al. Problems assessing uptake of Huntington disease predictive testing and a proposed solution. Eur J Hum Genet. 2009;17(1):66-70.
- Rodrigues FB, Ferreira JJ, Wild EJ. Huntington’s disease clinical trials corner: June 2019. J Huntingtons Dis. 2019;8(3):363-71.
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al; Phase 1–2a IONIS-HTTRx Study Site Teams. Targeting huntingtin expression in patients with Huntington’s disease. N Engl J Med. 2019;380(24):2307-16.
Engelsk sammanfattning
Huntington’s disease is an autosomal dominant neurodegenerative disease that leads to premature death. The disease is caused by a pathological CAG triplet expansion in the huntingtin gene. The disease is most common in Western populations, with onset in middle age and causing progressive motor, cognitive, and psychiatric symptoms. Currently, only symptomatic treatment is provided, but new molecular technologies may allow treatments reducing levels of mutated huntingtin.