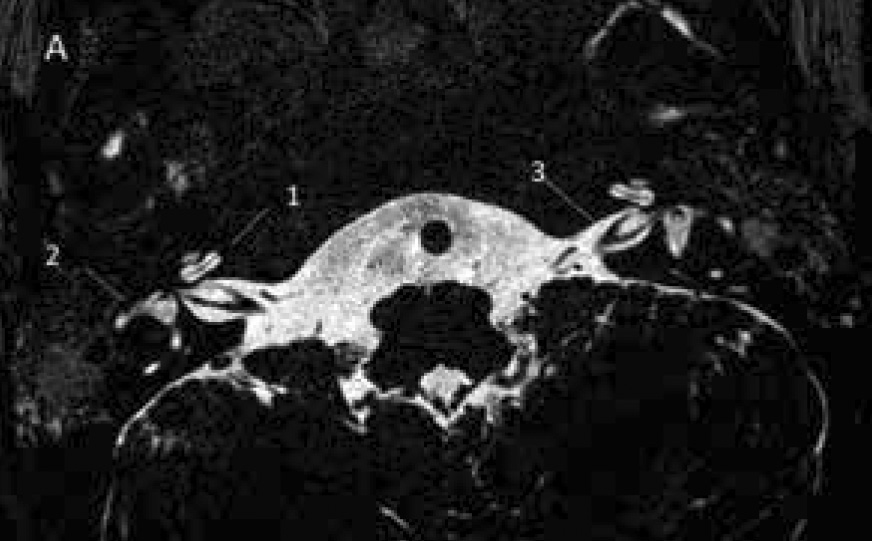

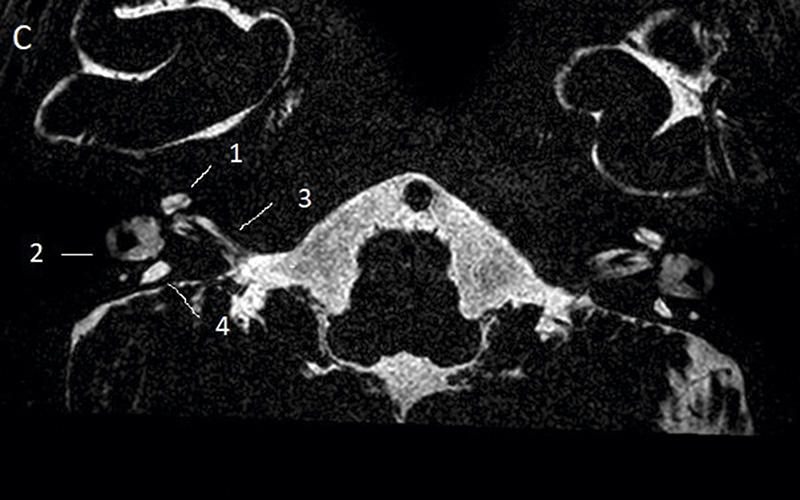
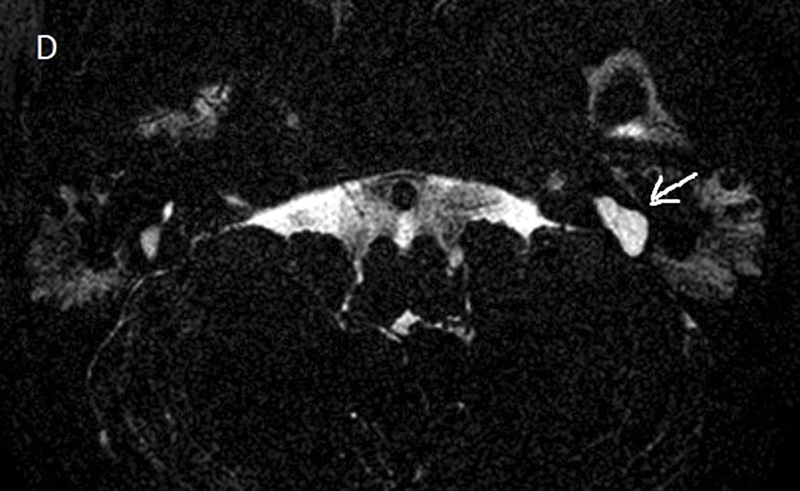
![Figur 3A. Normal anatomi. ant: anterior; po [?]: posterior; VII: facialisnerven i övre främre delen; cochlearis: n cochlearis i främre nedre delen; VS, VI: n vestibularis superior och inferior i bakre delen.](https://lakartidningen.se/wp-content/uploads/2020/09/Figur3awebb.jpg)

Hörselskadans orsak är bäst känd vid syndromal eller uttalad hörselnedsättning och dövhet.
Kongenital CMV-infektion kan orsaka medfödd, senare debuterande och progredierande hörselnedsättning, men även skador på balansorganet och kognitiva funktioner.
Majoriteten av genetiska hörselnedsättningar är lokaliserade till innerörat, och genetisk diagnos på medfödda hörselnedsättningar kan i dag göras i 60–70 procent av fallen, en andel som förväntas öka de närmaste åren.
Radiologi är numera en integrerad del av utredningen av barn med hörselnedsättning där anatomiska avvikelser bidrar till diagnos och bedömning av förutsättningar för kokleaimplantat.
Hörselnedsättning hos barn orsakas liksom de flesta sjukdomar av arv och miljö. Hos spädbarn handlar det i de flesta fall om ett i övrigt friskt barn med normalhörande föräldrar som efterfrågar en diagnos. Medfödd eller tidigt debuterande hörselnedsättning orsakas främst av genetiska faktorer (Figur 1), men även kongenitala infektioner och neonatala komplikationer (t ex grav asfyxi), behandling med ototoxiska läkemedel och meningit/encefalit [1]. Hörselnedsättning ingår i ett stort antal syndrom och kan ibland vara den första avvikelsen som upptäcks via hörselscreening av nyfödda.
Hörselnedsättning delas in i ledningshinder och sensorineurala hörselnedsättningar. Ledningshinder omfattar hörselgång och mellanöra, och de sensorineurala hörselnedsättningarna omfattar innerörat och hörselnerven. Permanent hörselnedsättning är oftast sensorineural och huvudsakligen lokaliserad till innerörat. Hörselskadan kan även omfatta hörselnerv och hjärnstam, t ex vid nervatresi och kärnikterus (svår neonatal hyperbilirubinemi). Ledningshinder eller konduktiv hörselnedsättning föreligger vid missbildning av hörselgång och/eller mellanöra, men kan även uppstå till följd av trauma eller kronisk infektion.
Bilateral hörselnedsättning ses oftast vid genetiska orsaker och neonatala komplikationer, medan ensidig hörselnedsättning är överrepresenterad vid kraniofaciala missbildningar [2]. Vid nytillkommet ledningshinder och normalt öronstatus kan orsaken vara ett kongenitalt kolesteatom, och detta bör därför utredas med röntgen.
Intrauterina infektioner, som rubella, cytomegalovirus (CMV) med flera agens, har också visats orsaka medfödd hörselnedsättning. I avsaknad av välfungerande vaccinationsprogram är kongenitalt rubellasyndrom fortfarande en vanlig orsak till medfödd hörselnedsättning, men i mer utvecklade områden har cytomegalovirus tagit över som den vanligaste intrauterina infektionen. Bakteriell meningit hos små barn orsakad av Haemophilus influenzae eller pneumokocker gav tidigare upphov till ett flertal fall av hörselskada och dövhet, men med utökat vaccinationsprogram är detta numera sällsynt.
Bilddiagnostik med MR och DT kan visa avvikelser i innerörat och hörselnerven som förklarar nedsatt funktion och även utgör en pusselbit i den genetiska utredningen. Prevalensen av hörselskadors etiologi relaterar främst till uttalad och syndromal hörselnedsättning, och fortsatta studier av måttlig, lätt och ensidig hörselnedsättning kan ändra bilden.
Kongenital CMV-infektion
Sambandet mellan kongenital CMV-infektion (cCMV) och hörselnedsättning observerades första gången 1964 [3] och i Sverige 1984 [4]. Kongenial CMV-infektion är i dag den vanligaste kända orsaken till medfödd icke-genetisk hörselnedsättning; den anses orsaka ca 20 procent av medfödd grav hörselnedsättning/dövhet, men kan även ge upphov till senare debuterande hörselnedsättning. Förekomsten av cCMV varierar mellan 0,2 och 2,5 procent av alla nyfödda och är högre i utvecklingsländer. Seroprevalensen hos mödrar ligger på 40–60 procent i Europa men >90 procent i utvecklingsländer. Viruset sprids via kroppsvätskor och den vanligaste smittkällan är barn i 1 till 2 års ålder [5]. cCMV orsakas framför allt vid primärinfektion, men kan förekomma även vid reinfektion eller reaktivering av tidigare infektion. Detta är en bidragande orsak till svårigheter att få fram ett effektivt vaccin [6].
Ungefär 10–15 procent av barnen med cCMV har generella, allvarliga symtom vid födseln. Eftersom merparten är symtomfria och CMV-screening av nyfödda saknas är omfattningen av cCMV-skador dåligt känd. Hörselnedsättning är vanligast förekommande, men även neurologiska symtom, balansrubbning och beteendeavvikelser har beskrivits [7]. Hörselnedsättningen kan vara både medfödd, senare debuterande eller progredierande så att en ensidig eller lätt hörselnedsättning utvecklas till en dubbelsidig, grav nedsättning. Vid konstaterad cCMV rekommenderas uppföljning av hörseln under minst sex år. Primärinfektion under den första trimestern verkar utgöra störst risk för fostret [8].
Diagnosen ställs på virusodling av urin inom 3 veckor från födseln. Men det är bara symtomatiska barn som provtas neonatalt. Hörselscreening sker oftast inom en vecka, men det kan ta flera veckor innan hörseldiagnosen är fastställd. CMV-antikroppar hos modern visar om det förelegat infektion under graviditeten. CMV-antikroppar hos äldre barn kan ha förvärvats postnatalt, och diagnosen kongenital CMV-infektion måste då fastställas med PCR-teknik avseende virus-DNA från PKU-lappen. Vid tidig diagnos kan behandling med valganciklovir ha en stabiliserande effekt på hörseln [9], vilket indikerar ett behov av CMV-screening av nyfödda.
Genetiska hörselnedsättningar
Genetiska faktorer orsakar 60–70 procent av hörselnedsättning hos barn, och man har i dag identifierat ett stort antal gener där en mutation är kopplad till hörselnedsättning. Syndromal hörselnedsättning uppges utgöra 30 procent av all genetiskt orsakad hörselnedsättning, och hörselundersökning ska därför alltid ingå i syndromutredning, särskilt vid multiorganpåverkan eller missbildning i ansikte–skallbas.
Hörselorganen anläggs mycket tidigt i fosterutvecklingen, vilket också innebär att ett stort antal gener (> 400) kan orsaka hörselnedsättning. Majoriteten av genetiska hörselnedsättningar är recessiva (80 procent) och lokaliserade till innerörat (90 procent), där mutationer i gener som styr uppbyggnad och jontransport anses vara den vanligaste orsaken. I dag finns mer än 100 icke-syndromala hörselgener lokaliserade. Utvecklingen går snabbt och aktuell information kan fås på https://hereditaryhearingloss.org.
Den vanligaste genetiska orsaken till hörselnedsättningen är mutation av connexin 26 (Cx 26, DFNB1A, GJB2). Ärftligheten är autosomalt recessiv och genen reglerar natrium–kaliumtransport. En annan mycket vanlig orsak är mutation av DFNB16 STRC (stereocilin). Denna hörselnedsättning kan komma att visa sig vara den vanligaste, då den kan uppkomma såväl prelingualt (före språkutveckling) som postlingualt [10]. Mutationer i mitokondrie-DNA kan ge upphov till hörselnedsättning, vanligen genom auditiv neuropati (hörselnervsskada). Ett annat exempel är A1555G-mutation, där miljö och genetik samverkar. Denna sjukdom ger försämrad metabolism av garamycin (gentamycin), vilket kan ge hörselnedsättning/dövhet vid behandling.
»Large vestibular aqueduct syndrome« (LVAS) innebär förstorad vestibulär akvedukt. Sjukdomen är autosomalt recessiv. Prevalensen är ca 10 procent av medfödd hörselnedsättning. Hörselnedsättningen kan vara lindrig–uttalad och är vanligen progressiv. Sannolikt är förstorad akvedukt inte huvudorsaken till hörselnedsättning utan ett bifynd. Några med LVAS har en genetisk mutation som orsakar Pendreds syndrom (SLC26A4) vilket innebär hypotyreos, vanligen debuterande i tonåren.
Personer med Treacher–Collins syndrom har kraniofaciala och otogena missbildningar. Ärftlighetsgången är autosomalt dominant med varierande penetrans. Ett annat exempel är brankiootorenalt syndrom (BOR), som ger laterala halsfistlar, missbildning av snäcka, labyrint (balans) och njurmissbildning/-dysfunktion, där hörselnedsättningen kan variera från måttlig till dövhet.
Den vanligaste syndromkombinationen är öga–öra. Det beror sannolikt på att dessa organ anläggs vid samma tid, har likartade strukturer och regleras av samma gener. Cirka 2 000 personer i Sverige har medfödd allvarlig grad av hörsel-/synnedsättning (dövblindhet). Exempel är Ushers, Alströms och Alports samt CHARGE-syndrom. Ushers syndrom är vanligast (8–10/100 000) och har tre kliniska typer. Typ 1 ger medfödd dövhet samt bilateral vestibulär areflexi med försenad grovmotorisk utveckling (gångdebut > 18 månader). Synnedsättning beror på näthinnesjukdomen retinitis pigmentosa (RP). Symtom är ökad ljuskänslighet, försämrat kontrastseende, obefintligt mörkerseende, kikarseende och katarakt. Typ 2 ger medfödd måttlig hörselnedsättning, normal vestibulär funktion och RP med liknande förlopp som vid typ 1. Typ 3 ger medfödd måttlig hörselnedsättning hos barn som förvärras och ger vuxendövhet; RP likartad som vid typ 1 och 2 [11].
Sedan kartläggningen av det mänskliga genomet har forskningsframstegen varit mycket stora. Exom eller helgenomsekvensering, som för 3–4 år sedan var mycket komplicerad, kan i dag göras mycket snabbt till en kostnad av 5 000–15 000 kronor. Till en mycket rimlig kostnad kan man i dag ställa korrekt klinisk och genetisk diagnos på medfödda hörselnedsättningar i ca 60–70 procent av fallen, en andel som förväntas öka de närmaste åren. Korrekt genetisk diagnos är en förutsättning för behandling. I dag pågår en stor mängd djurförsök och även humana genterapistudier (t ex vid Ushers syndrom). Behandlingstekniker som används är adenovirusvektorer, antisense-oligonukleotider och nya läkemedel (ataluren) samt Crispr. Nästa stora genombrott förväntas vara att med genterapi kunna behandla och stoppa progressiv hörselnedsättning.
Bilddiagnostik vid hörselnedsättning
Radiologi är numera en integrerad del av utredningen av barn med medfödd uttalad hörselnedsättning. DT och MR utförs i narkos hos små barn och kan med fördel samordnas med hörselundersökning med hjärnstamsaudiometri. Information om temporalbenets anatomi och eventuella missbildningar såväl i öronen som i övriga delar av skallen utgör nödvändigt underlag för bedömning av om patienten är lämplig för kokleaimplantat (CI) [12, 13].
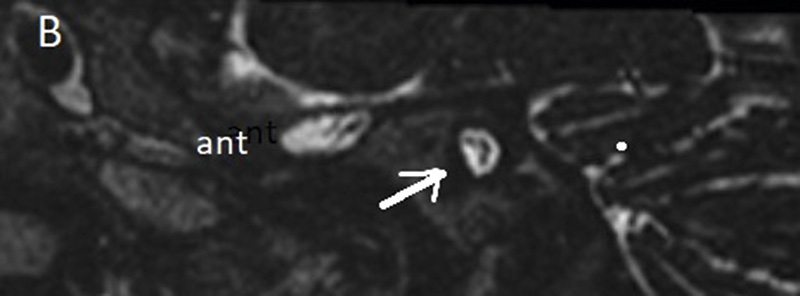
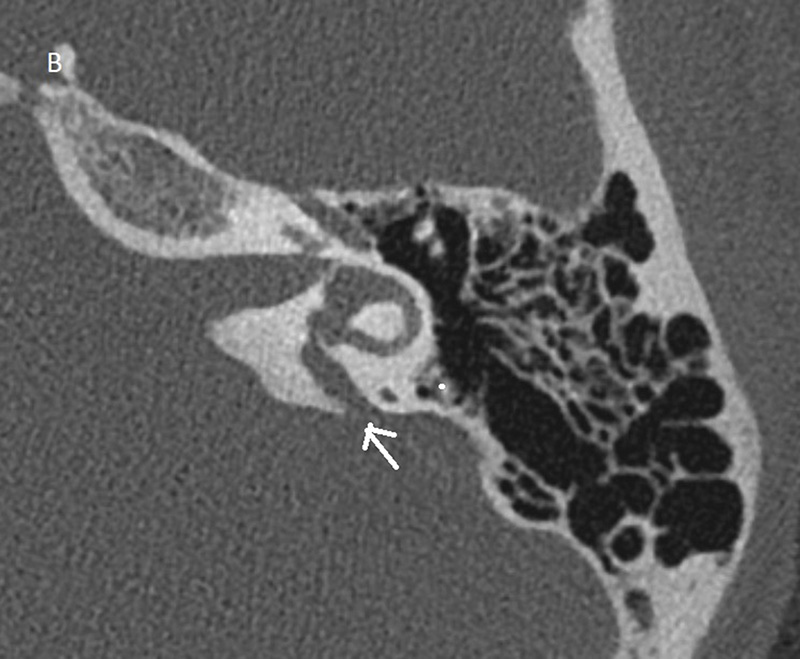
Temporalbenet är så litet att bildtagning med s k submillimetriska sekvenser är obligatorisk såväl med DT som MR [14]. DT avbildar ytter-, mellan- och innerörats beniga strukturer, medan högupplöst T2-viktad MR-sekvens med 3D-teknik möjliggör visualisering av innerörat och vestibulocochlearis-nerven. Kvalitetskraven är mycket höga, och man måste kunna identifiera lamina spiralis, scala tympani och vestibuli samt få skarpa bilder över facialis- och vestibulocochlearis-nerverna (Figur 2A–D, 3A,B samt 4A,B). Utredningen bör alltid kompletteras med avbildning av hjärnvävnaden för att kunna identifiera eller utesluta ytterligare anomalier och bedöma hörselbanorna.
Om frågeställningen gäller kartläggning av mellanörat vid t ex yttre hörselgångsatresi, och sensorineural hörselnedsättning inte föreligger, räcker DT för avbildning. Bedömning av missbildningarna sker med hjälp av Yeakleys och Jahrsdoerfers skala [15, 16]. Vid undersökning av barn med ensidig sensorineural hörselnedsättning är MR det bästa valet. I ca 30 procent av medfödda fall med ensidig sensorineural hörselnedsättning ser man missbildningar i innerörat. Missbildningar av inneröronen och vestibulocochlearisnerven klassificieras enligt Sennaroglu respektive Casselman [17-19].
Vid utredning inför kokleamplantat bör radiologen besvara
- om det är möjligt att lägga implantatet i snäckan
- hur vestibulocochlearisnervens cochlearisgren ser ut
- om det finns förändringar i hjärnvävnaden
- om det finns tecken på andra sjukdomar (t ex otit).
De flesta medfödda icke syndromala orsaker till sensorineurala hörselnedsättningar (rubella, alkohol, hypoxisk ischemisk skada) ger inte någon radiologiskt synlig förändring av innerörat. Inneröremalformation orsakad av cCMV är mycket varierande och nästan alltid associerad med anomalier i hjärnvävnaden [12]. Den vanligaste patologin hos barn med icke syndromal sensorineural hörselnedsättning är LVAS [12].
I ett fåtal syndrom med sensorineural hörselnedsättning ses avvikelser i innerörat, men dessa fynd är oftast karakteristiska för syndromet. Hos de flesta barn med syndrom finns det också uttalade missbildningar i ytter- och mellanörat. Öronmissbildningar är vanligast vid brankiootorenalt syndrom, CHARGE-syndrom, Pendreds och Waardenburgs syndrom och X-bunden »stapes gusher« [13].
Sammantaget förekommer hörselnedsättning tillsammans med de flesta olika organdysfunktioner, varför barn med hörselnedsättning bör genomgå frikostig kontroll av hjärta, njure, neurologi, metabolism och syn. En korrekt och tidigt ställd diagnos är förutsättningen för tidigt insatta åtgärder och undvikande av onödig oro.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
(uppdaterad 2022-09-15)
Referenser
- Smith RJ, Bale JF Jr,White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879-90.
- Howell JB, Appelbaum EN, Armstrong MF, et al. An analysis of risk factors in unilateral versus bilateral hearing loss. Ear Nose Throat J. 2019;98(6):330-3.
- Medearis DN Jr. Viral infections during pregnancy and abnormal human development. Am J Obstet Gynecol. 1964;90(Suppl):1140-8.
- Harris S, Ahlfors K, Ivarsson S, et al. Congenital cytomegalovirus infection and sensorineural hearing loss. Ear Hear. 1984;5(6):352-5.
- Goderis J, De Leenheer E, Smets K, et al. Hearing loss and congenital CMV infection: a systematic review. Pediatrics. 2014;134(5):972-82.
- Coppola T, Mangold JF, Cantrell S, et al. Impact of maternal immunity on congenital cytomegalovirus birth prevalence and infant outcomes: a systematic review. Vaccines (Basel). 2019;7(4):129.
- Karltorp E, Löfkvist U, Lewensohn-Fuchs I, et al. Impaired balance and neurodevelopmental disabilities among children with congenital cytomegalovirus infection. Acta Paediatr. 2014;103(11):1165-73.
- Faure-Bardon V, Magny JF, Parodi M, et al. Sequelae of congenital cytomegalovirus following maternal primary infections are limited to those acquired in the first trimester of pregnancy. Clin Infect Dis. 2019;69(9):1526-32.
- Kimberlin DW, Jester PM, Sánchez PJ, et al. Valganciclovir for symptomatic congenital cytomegalovirus disease. N Engl J Med. 2015;372(10):933-43.
- Vona B, Hofrichter MAH, Neuner C, et al. DFNB16 is a frequent cause of congenital hearing impairment: implementation of STRC mutation analysis in routine diagnostics. Clin Genet. 2015;87(1):49-55.
- Möller C. Deafblindness: living with sensory deprivation. Lancet. 2003;362(Suppl):S46-7.
- Huang BY, Zdanski C, Castillo M. Pediatric sensorineural hearing loss, part 1: Practical aspects for neuroradiologists. AJNR Am J Neuroradiol. 2012;33(2):211-7.
- Huang BY, Zdanski C, Castillo M. Pediatric sensorineural hearing loss, part 2: Syndromic and acquired causes. AJNR Am J Neuroradiol. 2012;33(3):399-406.
- Quirk B, Youssef A, Ganau M, et al. Radiological diagnosis of the inner ear malformations in children with sensorineural hearing loss. Br J Radiol. Epub 14 jun 2019. doi: 10.1259/bjro.20180050.
- Yeakley JW, Jahrsdoerfer RA. CT evaluation of congenital aural atresia: what the radiologist and surgeon need to know. J Comput Assist Tomogr. 1996;20(5):724-31.
- Jacob R, Gupta S, Isaacson B, et al. High-resolution CT findings in children with normal pinna or grade I microtia and unilateral mild stenosis of the external auditory canal. AJNR Am J Neuroradiol. 2015;36(1):176-80.
- Sennaroğlu L, Bajin MD. Classification and current management of inner ear malformations. Balkan Med J. 2017;34(5):397-411.
- Sennaroğlu L, Saatci I. A new classification for cochleovestibular malformations. Laryngoscope. 2002;112(12):2230-41.
- Casselman JW, Offeciers FE, Govaerts PJ, et al. Aplasia and hypoplasia of the vestibulocochlear nerve: diagnosis with MR imaging. Radiology. 1997;202(3):773-81.
Summary
Hearing impairment (HI) in children with a congenital or early onset is explained by genetic and environmental factors. Genetic factors are regarded as responsible for more than half of the cases of congenital HI and environmental factors are responsible for another 14-30%, but the etiology is still unknown in 20-40% [1]. Regarding the genetic causes, 30% are considered syndrome related, whereas 70% non-syndromic. Congenital infections and neonatal trauma may explain 20-30%. Without a well-run vaccination program, congenital rubella syndrome remains the most important cause of acquired infectious congenital HI, whereas in more developed areas, congenital cytomegalovirus (cCMV) has become the most common intrauterine infection. cCMV may cause 20% of congenital HI, but it is also responsible for late onset and progressive HI. Development, including advanced MRI, cellular biology and genetic analysis has within a few years provided diagnostic advancements in the etiology of HI.