Precisionsmedicin i form av specifik molekylär diagnostik och skräddarsydd behandling är sedan länge standard för flera hematologiska sjukdomar.
Vid mer komplexa hematologiska maligniteter är genpanelsekvensering i dag ett viktigt diagnostiskt verktyg och utgör grund för prognostisk bedömning och riktad behandling.
Genpanelsekvensering kan även påvisa ärftlig predisposition för vissa hematologiska sjukdomar.
Precisionsverktyg för analys av minimal kvarvarande sjukdom efter behandling förbättrar uppföljningen.
Det finns ett stort antal precisionsläkemedel för hematologiska sjukdomar som är godkända eller befinner sig i klinisk prövning.
I många avseenden har det hematologiska fältet lett utvecklingen inom precisionsdiagnostik och behandling. En anledning är att det har varit lätt att få tillgång till tumörmaterial för studier av genetiska markörer, en annan att tumörceller lätt har kunnat odlas och exponeras för olika substanser, vilket har stimulerat utvecklingen av nya läkemedel. En bidragande orsak till framgångsrik läkemedelsutveckling är också att hematopoetiska tumörer vanligtvis har relativt få mutationer, vanligen färre än 10, vilket gör att de ofta är lättare att angripa än mer komplexa cancerformer. Samtliga hematologiska maligniteter beskrivs i WHO-klassifikationen, och antalet undergrupper är i dag uppe i närmare 200 – och fler tillkommer vid varje utgåva [1].
Myeloiska maligniteter
Kromosomanalys har länge varit en självklar del av både diagnostik och prognostik vid myeloiska maligniteter. Exempel på sjukdomar definierade av translokationer mellan två kromosomer är akut promyelocytleukemi med t(15;17) och kronisk myeloisk leukemi med t(9;22) [2, 3]. Translokationerna ger upphov till nya fusionstranskript (PML/RARA vid akut promyelocytleukemi och BCR-ABL1 vid kronisk myeloisk leukemi), vilka orsakar leukemiutveckling. För båda tillstånden utvecklades specifika inhibitorer som stoppar leukemitillväxten; akut promyelocytleukemi kan i stor utsträckning botas med vitamin A-syra i kombination med arsenik, och kronisk myeloisk leukemi är i dag en sjukdom som till >90 procent hålls i schack av målstyrd behandling med tyrosinkinashämmare. Tidigt utvecklades också tekniker för att detektera JAK2-mutation vid polycythaemia vera, och även här kan sjukdomen behandlas – om än inte botas – med specifika JAK2-inhibitorer [4].
Att komma vidare från de tidiga framgångarna visade sig svårare än förväntat. Man upptäckte att de flesta myeloiska maligniteter är genetiskt heterogena och kännetecknas av 2 till ca 10 mutationer. Till skillnad från JAK2-mutationen, som företrädesvis uppstår i ett specifikt baspar, är de flesta mutationer oftast spridda över hela anlaget. Den första helgenomsekvenseringen av en leukemi publicerades 2008 och beskrev 8 mutationer [5]. Detta blev startskottet för en snabb utveckling av så kallad genpanelsekvensering, där ett begränsat antal gener analyseras delvis eller i sin helhet. Panelsekvensering är snabb och relativt billig och kan användas för att identifiera muterade cellpopulationer ner till ca 3–5 procents nivå av benmärgens celler.
Snart hade flera blodsjukdomar kartlagts med avseende på sina mutationer. Akut myeloisk leukemi och myelodysplastiskt syndrom bär på i snitt 2–3 olika mutationer eller kromosomavvikelser [6, 7]. Vissa mutationer, till exempel i NPM1 vid akut myeloisk leukemi, innebär bättre prognos, medan andra talar för sämre prognos. Genom genpanelsekvensering av stora välkaraktäriserade material har man beskrivit det molekylära landskapet, det vill säga vilka typer av mutationer och kombinationer som ses i olika subgrupper. Muterade gener indelas i undergrupper beroende på sin funktion i cellen. Gengrupper som oftast uppvisar mutationer är involverade i epigenetisk reglering, RNA-splitsning, cellsignalering och transkription. Genen TP53, »genomets väktare«, kan precis som vid solid cancer vara muterad vid akut myeloisk leukemi och myelodysplastiskt syndrom, vilket vanligtvis innebär dålig prognos [8].
De myeloproliferativa sjukdomarna polycythaemia vera och essentiell trombocytemi har ofta endast en av tre kända mutationer, medan myelofibros ligger någonstans mitt emellan. En tumregel är att prognosen är sämre ju fler mutationer och kromosomavvikelser sjukdomen bär på.
Genetisk karaktärisering är i dag en del av WHO-klassifikationen, och panelsekvensering utgör en viktig del av rutindiagnostik och prognostik. Genomic Medicine Sweden (GMS) har utvecklat en myeloisk genpanel som omfattar 195 gener, vilken nu börjar användas i hela Sverige inom rutindiagnostik (Figur 1), och arbete pågår för närvarande för att sammanställa resultaten efter den initiala valideringsrundan (GMS, opubl data). Baserat på panelsekvensering kan man vid vissa undergrupper av akut myeloisk leukemi undvika allogen stamcellstransplantation om initial cellgiftsbehandling fungerar, medan man för andra redan vid diagnos vet att transplantation är det bästa alternativet. Nya mer eller mindre specifika precisionsläkemedel har utvecklats och används i studier eller i rutin vid akut myeloisk leukemi. Exempel är hämmare för de epigenetiska regulatorerna IDH1 och IDH2 och tyrosinkinashämmare för FLT3.
Studier under senare år har visat att hematologiskt friska individer kan bära på mutationer i en liten andel av sina blodceller. Detta kallas »CHIP« (clonal hematopoiesis of indeterminate potential) [9]. CHIP består vanligen av en eller högst två mutationer, och andelen muterade celler är låg. Förekomsten av CHIP ökar med stigande ålder och har i några, men inte alla, studier visats öka risken för både framtida hematologiska maligniteter och kardiovaskulär sjukdom. Om man i forskningssyfte och med känsliga metoder letar efter mycket små kloner, 1–2 procent av benmärgens celler, finner man CHIP hos över 50 procent av personer över 70 år. I dagsläget finns inga internationella eller svenska riktlinjer för hur CHIP ska hanteras, men det finns en konsensus att små CHIP-kloner hos äldre individer inte behöver följas upp och att man inte heller aktivt ska undersöka hematologiskt friska individer med denna frågeställning. CHIP hos yngre personer ska följas i samråd med hematolog, men exakta rekommendationer saknas.
Lymfatiska maligniteter
För akut lymfatisk leukemi är genetiska analyser sedan länge en viktig del av diagnostik och behandlingsprotokoll för barn och vuxna. I det senaste protokollet för patienter upp till 45 år (»All together«) görs en rad olika genetiska analyser (kromosomanalys, fluorescent in situ-hybridisering [FISH], arrayer) vid diagnos för att identifiera riskmarkörer som ändrar behandlingsstrategin.
Vid behandlingskrävande kronisk lymfatisk leukemi utförs FISH-analys för ett antal genetiska markörer (bland annat 17p-deletion) och sekvensering av TP53-genen. Många laboratorier har bytt till NGS-analys (next generation sequencing) av TP53-genen, vilket ger högre känslighet. TP53-avvikelser är prediktiva markörer för riktad behandling då dessa patienter svarar dåligt på kemoimmunterapi och i stället ska behandlas med BTK (ibrutinib)- eller BCL2 (venetoclax)-hämmare [10]. Med introduktion av målriktade terapier har resistensmutationer påvisats, bland andra BTK-mutationer, vilka lätt kan upptäckas med genpanelanalys.
För myelom utförs också en panel av FISH-analyser för att påvisa kromosomavvikelser med prognostisk information, såsom 1q-överskott, 17p-deletion och t(4;14).
För den stora gruppen lymfomsjukdomar har den genetiska diagnostiken hittills omfattat FISH-analyser av vanliga translokationer, till exempel t(11;14) vid mantelcellslymfom, t(14;18) vid follikulärt lymfom och t(8;14) vid Burkittlymfom. Under senare år har det molekylära landskapet karakteriserats i mer detalj, och ett antal gener har visats ha diagnostisk, prognostisk och prediktiv betydelse [11]. I rutindiagnostik finns genmutationer som förbättrar klassificeringen av lymfom, till exempel MYD88-mutationer vid Waldenströms makroglobulinemi, BRAF-mutationer vid hårcellsleukemi och RHOA, TET2 och IDH2 vid T-cellslymfom. Prognostiskt betydelsefulla genmutationer omfattar bland andra TP53 och NOTCH1-mutationer vid mantelcellslymfom och TP53-mutationer och MYC-translokationer vid diffust storcelligt B-cellslymfom. Därtill finns mutationer som har betydelse för svar på målinriktad terapi: om till exempel en Waldenström-patient har en MYD88-mutation utan CXCR4-mutation så är svaret på BTK-inhibitor bättre än vid samtidig MYD88- och CXCR4-mutation, eller om båda generna är omuterade [12].
För många lymfom vore det även önskvärt att kunna detektera strukturella avvikelser (till exempel translokationer) och mer komplexa analyser, såsom B-cell/T-cellreceptor-rearrangemang. Analys av IGHV-mutationsstatus utgör också en viktig prognostisk och sannolikt även prediktiv markör vid kronisk lymfatisk leukemi [13]. Inom GMS utvecklas nu en bred lymfatisk genpanel som omfattar analys av 252 gener (Figur 1), där nästa steg även inkluderar strukturella avvikelser och B-cell-/T-cellreceptor-rearrangemang. Förhoppningsvis kan man då göra en enda och mer omfattande panelsekvensering.
Benmärgssvikt och ärftlig predisposition
Kunskapen om att vissa ärftliga sjukdomar med debut under barndomen, som Fanconis anemi, Shwachman–Diamonds syndrom och Diamond–Blackfans anemi, kan leda till akut myeloisk leukemi och myelodysplastiskt syndrom har funnits länge. Däremot utgick man ifrån att nästan alla myeloiska sjukdomar i vuxen ålder inte var kopplade till genetisk predisposition. År 1999 upptäcktes ärftliga mutationer i transkriptionsfaktorn RUNX1 som är associerade med lätt trombocytopeni och ökad risk för utveckling av akut myeloisk leukemi eller myelodysplastiskt syndrom. Sedan dess har ett flertal ärftliga mutationer associerats med myeloiska maligniteter [14]. Som vid andra ärftliga sjukdomar varierar penetransen, det vill säga hur stor andel av mutationsbärare som utvecklar sjukdom [15]. Detta kan innebära att ärftliga sjukdomar kan »hoppa över« en generation, det vill säga att patienter som bär på anlaget inte utvecklar sjukdomen. Detta är viktigt att ta hänsyn till vid ärftlighetsanamnes. Man uppskattar att omkring 50 procent av generna som är kopplade till genetisk predisposition för myeloiska maligniteter är kända i dag. GMS myeloida genpanel innehåller de vanligaste predispositionsmutationerna, och en nationell studie pågår för att undersöka prevalens, penetrans och expressivitet för kända ärftliga syndrom och samtidigt identifiera »nya« mutationer.
WHO-klassifikationen inkluderar i dag 10 tillstånd med genetisk predisposition för myeloiska maligniteter [1], vilka uppskattas ligga bakom 1–10 procent av all myeloisk cancer, med högre incidens hos unga patienter. En studie visade till exempel att ärftliga GATA2-mutationer finns hos 12 procent av patienter med myelodysplastiskt syndrom yngre än 45 år men hos bara 2 procent av de som är äldre än 45 år [16].
Att identifiera patienter med genetisk predisposition är viktigt för val av behandling. Vid till exempel Fanconis anemi och telomersjukdomar ökar känsligheten för cytostatika betydligt, vilket medför risk för cytostatikarelaterad mortalitet, inte minst i samband med stamcellstransplantation. Vid autosomalt dominanta mutationer måste man dessutom beakta en 50-procentig risk att ett HLA-identiskt syskon är bärare av samma mutation och att en obesläktad donator då är att föredra.
För att underlätta klinisk handläggning och utveckling inom området har specialister från Norden utformat de första rekommendationerna om hur man ska diagnostisera, följa och behandla patienter med genetisk predisposition för myeloida maligniteter [15].
Minimal kvarvarande sjukdom
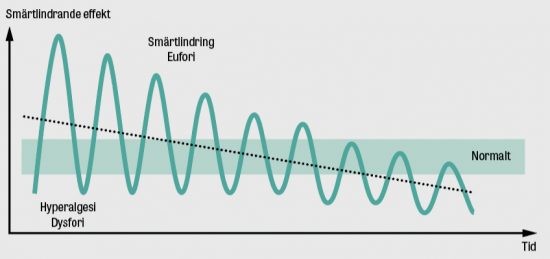
Minimal kvarvarande sjukdom (MRD, minimal [measurable] residual disease) motsvarar andelen maligna celler som inte kan ses under mikroskopet men som finns kvar efter behandling och kan leda till återfall av sjukdomen (Figur 2). På senare år har nya känsliga tekniker utvecklats för att kunna påvisa MRD hos patienter i morfologisk remission, vilket gör att behandlingen kan anpassas för att förbättra patientens överlevnad. Vid akut leukemi kan MRD-positivitet leda till en mer intensiv behandling eller beslut om stamcellsstransplantation [17]. Dagens MRD-metoder baseras på flödescytometri eller på molekylära analyser av DNA och RNA [18, 19].
Flödescytometri detekterar en kombination av olika antigener som uttrycks på leukemicellerna vid diagnos som sedan kan identifieras efter behandling. Det prognostiska och prediktiva värdet av flödescytometri varierar mellan diagnosgrupper. Vid till exempel akut myeloisk leukemi kan flödescytometri identifiera en leukemisk cell bland 1 000–10 000 friska celler. Fynd av mer än 0,1 procent leukemiska celler innebär en högre risk för återfall och kan utgöra en indikation för stamcellstransplantation. Däremot har metoden ett lågt negativt prediktionsvärde för att utesluta återfall av akut myeloisk leukemi [19].
Fusionstranskript detekteras ofta på RNA-nivå med kvantitativ RT-PCR. Förutom BCR-ABL1 vid kronisk myeloisk leukemi och Philadelphia-positiv akut lymfatisk leukemi används i dag ett flertal fusionstranskript som MRD-markörer i klinisk rutin, med en känslighet ner till 1/100 000 transkript. En nackdel är att endast en mindre andel av patienter med hematologiska maligniteter uppvisar kända fusionstranskript i tumörcellerna [18, 19]. En relativt ny metod, RNA-sekvensering, kan påvisa ovanliga fusionsgener vid diagnos, vilket kan användas för att utveckla en »personlig« MRD-analys för uppföljning.
Specifika MRD-test kan också utformas för punktmutationer eller mindre insertioner/deletioner som hittas vid diagnos. Dessa förändringar kan påvisas på DNA- eller RNA-nivå med kvantitativ PCR eller digital PCR, med en något lägre känslighet än för fusionstranskript. Vid akut myeloisk leukemi används i dag kvantitativ PCR för uppföljning av NPM 1-muterade blodsjukdomar. En ny utveckling är digitala PCR-test som designas för individuella mutationer hos enskilda patienter. Vidare kan genpaneler användas som MRD-test, vilket trots en lägre känslighet (2–3 procent) har fördelen att ge en bättre bild av den klonala utvecklingen under behandling och återfall [20].
Vid B-/T-akut lymfatisk leukemi uppvisar varje tumör unika immunoglobulin- och T-cellreceptor-rearrangemang som kan användas som MRD-markörer [18]. Vid diagnos kan man identifiera de leukemispecifika sekvenserna med hjälp av sekvensering. Därefter designas patientspecifika PCR-reagenser som kan detektera en muterad molekyl bland 100 000 normala [18]. Metoden har standardiserats inom EuroMRD-konsortiet för vuxna och barn med akut lymfatisk leukemi och används vid riskgruppering och behandlingsval.
Framtida metodutveckling
Den metodologiska utvecklingen inom hematologiska maligniteter har varit snabb och i många fall vägledande för andra cancerformer. Dagens diagnostiska arsenal kräver dock stora expertkunskaper med avseende på metoder och tolkning. Inom GMS pågår i dag ett arbete inom akuta leukemier där helgenomsekvensering kombineras med RNA-sekvensering för att se om dessa tekniker kan ersätta nuvarande analysmetoder (Figur 1). Helgenomanalys kan påvisa mutationer i samtliga gener och samtidigt ge en bild av för WHO-klassifikationen viktiga kopietals- och strukturella förändringar, och skulle på sikt kunna ersätta kromosomanalys, FISH-analyser, riktade analyser samt breda genpanelanalyser. Likaså kan RNA-sekvensering upptäcka genfusioner som i dag analyseras med riktade analyser. Reagenskostnaderna för att utföra helgenomanalys och RNA-sekvensering är höga, även om de minskar i snabb takt, och metoden kommer att behöva genomgå en betydande analytisk och klinisk validering inför klinisk implementering. Analyser av hälsoekonomiska aspekter avseende dessa nya analyser pågår också inom GMS.
RNA-sekvensering kan också mäta uttrycket av alla gener i tumörcellerna, vilket kan ge värdefull diagnostisk och prognostisk information [21]. Även om denna kunskap inte är rutin i dag är det troligt att genuttrycksmönster, precis som vid bröstcancer, kommer att bli allt viktigare inom blodcancer [21, 22]. Slutligen har en helt ny teknologi utvecklats som gör det möjligt att studera molekylära förändringar i enskilda celler. Med så kallad singelcellanalys kan man studera tusentals enskilda celler på genetisk, epigenetisk och transkriptionell nivå och därmed få en uppfattning om heterogeniteten bland tumörcellerna, påverkan på den normala blodbildningen och vad som kännetecknar kvarvarande och resistenta tumörceller [23, 24]. I takt med att singelcellteknologin standardiseras, kostnaderna minskar och metoder anpassas för kliniskt bruk kan den komma att inta en framträdande plats i den diagnostiska arsenalen.
Sammanfattning
Vi upptäcker allt fler kliniskt relevanta genetiska förändringar vid blodcancer, och nya metoder och algoritmer utvecklas som leder till förfinad diagnostik och förbättrad prognosbedömning och val av behandling för den enskilde patienten. Med utvecklingen mot precisionsbehandling följer behovet att snabbt kunna komplettera nuvarande genetiska analyser för att påvisa specifika mutationer, anpassa behandling och följa sjukdomen med känsliga MRD-analyser. Samtidigt pågår en intensiv utveckling av nya metoder och lovande precisionsläkemedel som på ett avgörande sätt kommer att förbättra prognosen för ett flertal patientgrupper.
Potentiella bindningar eller jävsförhållanden: Samtliga författare är medlemmar i arbetsutskottet för hematologi inom Genomic Medicine Sweden (GMS). P-O Andersson har deltagit i rådgivande kommitté för AbbVie och Janssen. Thoas Fioretos är en av grundarna och tillika styrelseledamot i företagen Qlucore AB och Cantargia AB. Richard Rosenquist Brandell har deltagit i rådgivande kommittéer för AbbVie och Illumina.
Fakta 1. Kliniska exempel
- 70-årig kvinna med kronisk lymfatisk leukemi behandlades 2013 med kemoimmunterapi med god effekt. Våren 2020 återkom sjukdomssymtom, och inför start av behandling genomfördes mutationsanalyser som visade dels en nytillkommen TP53-mutation, dels en omuterad IGHV-gen. Dessa markörer indikerar att kemoimmunoterapi kommer att vara otillräcklig: ger sämre svar, betydligt kortare duration samt en ökad immunhämning. I stället kan en riktad behandling ges i form av BTK-hämmare (till exempel ibrutinib), som fungerar bra vid båda dessa förändringar.
- 73-årig i övrigt frisk man diagnostiserades 2018 med asymtomatisk pancytopeni. Diagnosen blev lågrisk myelodysplastiskt syndrom och det fanns ingen indikation för stamcellstransplantation. Panelsekvensering visade två patogena mutationer som bekräftade myelodysplastiskt syndrom men som saknade negativ prognostisk betydelse. 2019 sågs ny klon inom RAS-signalvägen på 5 procent, och man kunde då påvisa den i föregående prov på 2 procents nivå. 2020 hade cytopenin aggraverats, morfologin förvärrats och den nya klonen ökat till 29 procent. Utvecklingen mot progressivt myelodysplastiskt syndrom med dålig prognos, samt att patientens allmäntillstånd var fortsatt gott, gjorde att multiprofessionell konferens tog beslut om att erbjuda stamcellstransplantation.
(uppdaterad 2021-05-11)
Referenser
- Swerdlow SH, Campo E, Harris NL, et al (editors). WHO Classification of tumours of haematopoietic and lymphoid tissues. Revised 4th ed. Lyon: International Agency for Research on Cancer (IARC); 2017.
- Hochhaus A, Larson RA, Guilhot F, et al; IRIS Investigators. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917-27.
- Sanz MA, Fenaux P, Tallman MS, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133(15):1630-43.
- McMullin MF, Harrison CN, Ali S, et al; BSH Committee. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Br J Haematol. 2019;184(2):176-91.
- Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66-72.
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-21.
- Papaemmanuil E, Gerstung M, Malcovati L, et al; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616-27; quiz 99.
- Bernard E, Nannya Y, Hasserjian RP, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26(10):1549-56.
- Libby P, Sidlow R, Lin AE, et al. Clonal hematopoiesis: crossroads of aging, cardiovascular disease, and cancer: JACC review topic of the week. J Am Coll Cardiol. 2019;74(4):567-77.
- Campo E, Cymbalista F, Ghia P, et al. TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. 2018;103(12):1956-68.
- Rosenquist R, Rosenwald A, Du MQ, et al; European Research Initiative on CLL (ERIC) and the European Association for Haematopathology (EAHP). Clinical impact of recurrently mutated genes on lymphoma diagnostics: state-of-the-art and beyond. Haematologica. 2016;101(9):1002-9.
- Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372(15):1430-40.
- Rosenquist R, Ghia P, Hadzidimitriou A, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: updated ERIC recommendations. Leukemia. 2017;31(7):1477-81.
- Godley LA, Shimamura A. Genetic predisposition to hematologic malignancies: management and surveillance. Blood. 2017;130(4):424-32.
- Baliakas P, Tesi B, Wartiovaara-Kautto U, et al. Nordic guidelines for germline predisposition to myeloid neoplasms in adults: recommendations for genetic diagnosis, clinical management and follow-up. Hemasphere. 2019;3(6):e321.
- Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376(6):536-47.
- Gilleece MH, Labopin M, Yakoub-Agha I, et al. Measurable residual disease, conditioning regimen intensity, and age predict outcome of allogeneic hematopoietic cell transplantation for acute myeloid leukemia in first remission: a registry analysis of 2292 patients by the Acute Leukemia Working Party European Society of Blood and Marrow Transplantation. Am J Hematol. 2018;93(9):1142-52.
- Della Starza I, Chiaretti S, De Propris MS, et al. Minimal residual disease in acute lymphoblastic leukemia: technical and clinical advances. Front Oncol. 2019;9:726.
- Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131(12):1275-91.
- Sánchez R, Ayala R, Martínez-López J. Minimal residual disease monitoring with next-generation sequencing methodologies in hematological malignancies. Int J Mol Sci. 2019;20(11):2832.
- Cieslik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet. 2018;19(2):93-109.
- Haferlach T, Schmidts I. The power and potential of integrated diagnostics in acute myeloid leukaemia. Br J Haematol. 2020;188(1):36-48.
- Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet. 2019;20(5):257-72.
- van Galen P, Hovestadt V, Wadsworth Ii MH, et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176(6):1265-81.e24.
Summary
Precision diagnostics and therapy have been implemented rather early in clinical hematology due to the easy accessibility of blood and bone marrow, allowing not only for consecutive genetic analysis at diagnosis, remission and relapse, but also for culturing these cells and testing new drugs in vitro. One contributing factor has also been the relatively low number of »driver« mutations in hematologic malignancies and that some of them are gain of function mutations that are relatively easy to target by drugs. Examples of this development are ABL1-, JAK2-, and FLT3-inhibitors for the treatment of chronic myeloid leukemia, myeloproliferative neoplasms, and acute myeloid leukemia, respectively. More recently, gene panel sequencing has been introduced in clinical routine to identify genetic alterations with diagnostic, prognostic and predictive impact, and more sensitive techniques to monitor minimal residual disease are emerging. Whole genome and transcriptome sequencing are currently evaluated as the next diagnostic tool. Finally, a large number of targeted therapies are currently under development and/or undergoing clinical trials.