Pulmonell arteriell hypertension är en allvarlig sjukdom där förbättrad diagnostik och förfinad behandling bidragit till att förbättra patienternas prognos och funktionsförmåga.
Svensk förening för pulmonell hypertension (SveFPH) arbetar för en nationell samsyn, kvalitetsregister och multiprofessionellt omhändertagande.
Patienter med pulmonell arteriell hypertension är vanligare än man tidigare uppfattat och medelåldern ligger högre än förväntat, men i nivå med andra internationella register.
Drygt 40 procent av patienterna behandlas med fler än ett för pulmonell arteriell hypertension specifikt läkemedel, vilket förväntas öka och initieras redan i ett tidigt skede.
För fem år sedan publicerades en översiktsartikel om pulmonell arteriell hypertension i Läkartidningen [1]. Sedan dess har mycket hänt inom diagnostik och behandling. Diagnosen pulmonell arteriell hypertension är inte fullt så ovanlig som tidigare antagits, medelåldern är högre än förväntat och modern behandling har förbättrat överlevnaden för de drabbade patienterna. Medianöverlevnaden för obehandlade patienter med idiopatisk pulmonell arteriell hypertension anses vara 2,8 år [2], det vill säga knappt 3 år. Överlevnaden har förbättrats i takt med att nya behandlingar har tagits fram. I den senaste rapporten från det svenska registret för pulmonell arteriell hypertension (SPAHR) var 3-årsöverlevnaden för patienter med idiopatisk pulmonell arteriell hypertension cirka 75 procent [3] och medianöverlevnaden nästan 7 år.
Detta visar att prognosen är fortsatt allvarlig, vilket i kombination med att diagnostiken är komplicerad gör att patienterna ska handläggas av, eller i samråd med, centra för pulmonell arteriell hypertension vid landets universitetssjukhus där multidisciplinära team finns organiserade för denna verksamhet. Ytterligare skäl för centrering till universitetssjukhusen är den komplicerade och kostsamma behandlingen.
Vi presenterar här gällande rekommendationer för diagnostik och behandling samt resultat från det svenska registret för pulmonell arteriell hypertension.
Klassifikation och diagnostik
Det 5:e världssymposiet om pulmonell hypertension hölls i Nice i februari 2013. Här diskuterades området utifrån etiologi, genetik, klassifikation, diagnostik, evidensgrundad behandling och aktuella forskningsfronter. Vid mötet togs en modifierad klassifikation av pulmonell hypertension fram [4, 5] som skiljer sig något från den föregående (Dana Point 2008), se Fakta 1. Av dessa är det pulmonell hypertension sekundär till vänsterkammarsvikt, grupp 2, och pulmonell hypertension sekundär till lungsjukdomar såsom KOL och lungfibros, grupp 3, som är vanligast förekommande. Grupperingen syftar till att skilja mellan underliggande orsaker till pulmonell hypertension och att styra behandling. Grupp 1, pulmonell arteriell hypertension, utgör endast en mindre andel av patienterna och består av patienter med oförklarad, det vill säga idiopatisk pulmonell arteriell hypertension, patienter med hereditär pulmonell arteriell hypertension samt de med associerad pulmonell arteriell hypertension. I gruppen associerad pulmonell arteriell hypertension ingår de med bindvävssjukdom, främst sklerodermi, och de med kongenital hjärtsjukdom. Behandlingsalternativen som diskuteras nedan gäller endast för pulmonell arteriell hypertension.
Patobiologi
All pulmonell hypertension har någon grad av vaskulopati i lungkärlen där de gemensamma dragen är mediahypertrofi och glatt muskelhyperplasi i muskulära lungartärer, dilatation med plackbildning i elastiska artärer samt hypertrofi av höger kammare. Utmärkande för pulmonell arteriell hypertension är dessutom oblitererande intimaförtjockning med endotelcellsproliferation och fibros i prekapillära och intraacinära små artärer samt in situ-trombos [6]. I tillståndet ingår ett element av vasokonstriktion som förr ansågs drivande för sjukdomsprogressen. Numer bedöms den oblitererande effekten ha större betydelse för utveckling av förhöjd lungkärlsresistans [7].
Symtom och utredning
De vanligaste symtomen vid pulmonell arteriell hypertension är andfåddhet, särskilt vid ansträngning, trötthet, hjärtklappning, och ibland bröstsmärtor och svimning. Symtomen är gemensamma med många andra sjukdomar i hjärta och lungor. Utredningen blir därför omfattande för att korrekt diagnostisera och skilja pulmonell arteriell hypertension från övriga former av pulmonell hypertension. Ett väl strukturerat upplägg innehåller en god anamnes och en noggrann undersökning av patientens status, blodprov såsom Hb och NT-proBNP (N-terminal brain natriuretic peptide), vilo-EKG, spirometri med diffusionskapacitet, röntgenundersökning av lungorna, ekokardiografi, arbets-EKG, lungskintigrafi, högupplöst datortomografi av lungorna samt hjärtkateterisering. En objektiv värdering av funktionsförmågan görs oftast med ett 6-minuters gångtest. I utvalda fall görs konventionell pulmonalisangiografi och magnetkameraundersökning.
Ekokardiografisk undersökning av hjärtat och central cirkulation identifierar patienter med pulmonell hypertension, men för att ställa diagnosen pulmonell arteriell hypertension krävs hjärtkateterisering för bestämning av tryck och flöden i lungkretsloppet. Såväl diagnostiken som graderingen av sjukdomens svårighetsgrad baseras på en definition utgående från det uppmätta medelartärtrycket i lungartären, som ej fås fram från ekokardiografi. Medelartärtrycket från lungartären ligger vanligen under 20 mm Hg i vila, och pulmonell hypertension definieras som patologiskt vid ett medeltryck på 25 mm Hg eller mer. För definitiv värdering av pulmonell hypertension fordras bestämning av inkilningstrycket i lungartären, tidigare benämnt PCWP (pulmonary capillary wedge pressure) och numera PAWP (pulmonary arterial wedge pressure), eftersom det är lungartärens och inte kapillärens inkilningstryck som mäts. Termerna pre- (PAWP ≤15 mm Hg) respektive postkapillär (PAWP >15 mm Hg) pulmonell hypertension har införts och syftar till att skapa väldefinierade patientpopulationer, vilket påverkar terapivalet. En patient med postkapillär pulmonell hypertension har ett medelartärtryck i lungartären ≥25 mm Hg och ett PAWP >15 mm Hg, det vill säga vänstersidig hjärtsjukdom, och tillhör grupp 2 i klassifikationen, se Fakta 1. Patienter med pulmonell hypertension tillhörande grupp 1, 3, 4 eller 5 har en prekapillär pulmonell hypertension; det vill säga medelartärtryck i lungartären ≥25 mm Hg och PAWP ≤15 mm Hg. Det är värt att notera att definitionen för pulmonell hypertension om medelartärtryck i lungartären >30 mm Hg under arbete har utgått.
Även om pulmonell vaskulär resistans ej inkluderas i definitionen av pulmonell hypertension vid 5:e världssymposiet i Nice, så är det av värde för bedömning av den enskilde patienten att bestämma pulmonell vaskulär resistans vid hjärtkateterisering, då höger kammares afterload påverkar dess funktion och därmed det kliniska förloppet vid pulmonell hypertension [4]. Pulmonell vaskulär resistens infördes ånyo då de nya internationella riktlinjerna definierades 2015 [8].
Nationellt kvalitetsregister
Svensk förening för pulmonell hypertension (SveFPH) är en ideell sammanslutning av olika yrkesgrupper såsom läkare, sjuksköterskor, sjukgymnaster, biomedicinska analytiker och andra med intresse av tillståndet. Medlemmarna representerar inte enbart olika yrkesgrupper utan också olika specialiteter inom den moderna sjukvården. Föreningen bildades år 2007 och har bland annat som uppgift att inhämta och sprida kunskap i såväl sjukvården som i samhället kring pulmonell hypertension. Föreningen är remissinstans i frågor rörande pulmonell arteriell hypertension, arrangerar regelbundna vetenskapliga möten samt delar ut forsknings- och resestipendier. Sedan år 2008 driver Svensk förening för pulmonell hypertension det nationella kvalitetsregistret SPAHR, som drivs med ekonomiska medel från Sveriges Kommuner och landsting och vars registerplattform förvaltas av Uppsala Clinical Research Center. För ytterligare information om föreningens verksamhet, se www.svefph.se.
I Sverige finns vid universititetssjukhusen sju centra för pulmonell arteriell hypertension vilka alla rapporterar till SPAHR. Registret inkluderar patienter med pulmonell arteriell hypertension, kronisk tromboembolisk pulmonell hypertension och pulmonell hypertension av oklara och/eller multifaktoriella orsaker, det vill säga grupp 1, 4 och 5 i Niceklassifikationen (Fakta 1). Även patienter med pulmonell hypertension vid vänstersidig hjärtsjukdom och pulmonell hypertension vid lungsjukdom och/eller hypoxi (grupp 2 och 3 i klassifikationen) registreras om de erhållit behandling med läkemedel specifika för pulmonell arteriell hypertension på grund av en reaktiv oproportionerlig pulmonell arteriell hypertension. De sistnämnda patienterna, grupp 2 och 3 med läkemedel specifika för pulmonell arteriell hypertension, utgör endast ett fåtal av patienterna med dessa diagnoser och inkluderas i registret i enlighet med Socialstyrelsens riktlinjer [9], då detta är att betrakta som forskning och utveckling. SPAHR:s årsrapport ger en bild av sjukdomens incidens och prevalens, fördelning av diagnoser och behandling samt kliniska mått och överlevnad [3]. Resultaten visas för hela landet, men jämför också landets centra för pulmonell arteriell hypertension med varandra utifrån både processmått och behandlingsval. Drygt 85 procent av alla patienter med behandling specifik för pulmonell arteriell hypertension i Sverige är registrerade i SPAHR.
Prevalens och incidens
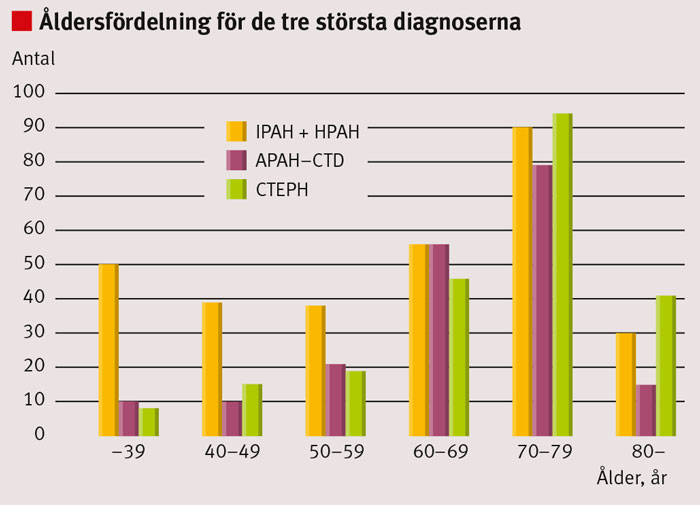
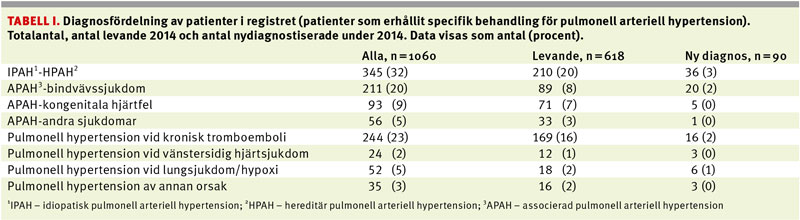
Det svenska registret innehöll den 31 december 2014 data från 1 060 patienter, varav 637 kvinnor och 423 män, med en medelålder på 59±17 år vid diagnos [3]. Två tredjedelar var diagnostiserade med pulmonell arteriell hypertension (n = 705; 65 procent kvinnor, medelålder 57±18 år) och en fjärdedel med pulmonell hypertension vid kronisk tromboemboli (n = 244; 49 procent kvinnor, medelålder 65±13 år). Liksom tidigare ses en dominans av kvinnor vid idiopatisk pulmonell arteriell hypertension men med en högre medelålder än förväntat, emellertid i överensstämmelse med internationella register [10, 11]. Mer detaljerade uppgifter om antalet patienter i de olika diagnosgrupperna framgår av Tabell I och åldersfördelning vid diagnos i Figur 1.
Vid utgången av år 2014 levde 618 av ovanstående patienter (377 kvinnor och 241 män), och 90 nya fall hade registrerats under året. Pulmonell arteriell hypertension har länge betraktats som en mycket ovanlig sjukdom med låga incidens- och prevalenstal, och registerstudier talar för prevalenstal på minst 15/miljon invånare och incidenstal omkring 10/miljon invånare [10, 11]. Motsvarande siffror för pulmonell arteriell hypertension i Sverige i dag är 25 och 5/miljon invånare för respektive prevalens och incidens (Tabell I).
Behandling
Tre basala mekanismer tycks bidra till de förändringar som leder till förhöjd pulmonell vaskulär resistans, vilket i sin tur leder till belastning av högerhjärtat: vasokonstriktion, proliferation av kärlväggen i små pulmonalisartärer samt in situ-trombotisering [12, 13].
Endoteldysfunktion förefaller ha en central roll i denna utveckling med markerade förändringar i lokala endotelin-, kvävemonoxid- (NO) och prostacyklinsystem [12]. Dessa förändringar utgör basen för de behandlingar för pulmonell arteriell hypertension som används i dag [13]. Mekanismerna tycks emellertid endast delvis förklara de kärlförändringar som uppstår, och ytterligare cellulära dysfunktioner har beskrivits [14]. Studier av nya behandlingsprinciper baserade på forskningsrön kring andra cellulära avvikelser pågår [15].
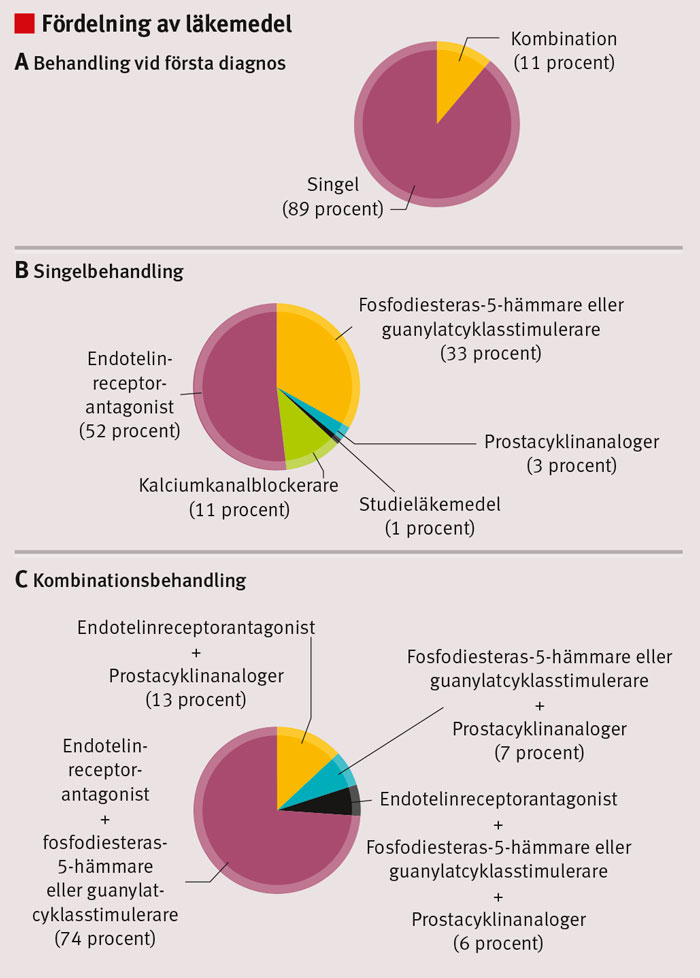
De farmakologiska behandlingsprinciperna för pulmonell arteriell hypertension följer de förändringar som beskrivs ovan genom 3 huvudlinjer; behandling med endotelinreceptorantagonister, fosfodiesteras-5-hämmare (PDE-5i) och prostacyklinanaloger. Det finns i dag rekommendationer om att kombinera dessa farmakologiska principer redan vid behandlingsstart [16]. I AMBITION-studien visades för första gången att initial kombinationbehandling var fördelaktig jämfört med singelbehandling avseende tid till klinisk försämring [17]. Av den svenska patientgruppen behandlades 11 procent med mer än ett för pulmonell arteriell hypertension specifikt läkemedel vid behandlingsstart (Figur 2) och 36 procent vid senast registrerade behandling [3]. Det vanligaste läkemedlet vid initiering av singelbehandling är endotelinreceptorantagonister, tätt följt av PDE-5i. Den vanligaste kombinationsbehandlingen är PDE-5i tillsammans med endotelinreceptorantagonister.
Flera studier finns gällande nya och uppdaterade behandlingsprinciper vid pulmonell arteriell hypertension [18, 19]. Effekterna av de nya läkemedlen/behandlingsprinciperna är tydliga, och läkemedlen kommer att utgöra ett värdefullt tillskott till den i dag etablerade behandlingen. Macitentan (Opsumit) är en ny dubbel endotelinreceptorblockerare, medan riociguat (Adempas) är en helt ny typ av läkemedel, guanylatcyklasstimulerare. Den sistnämnda verkar genom att direkt stimulera lösligt guanylatcyklas oberoende av kvävemonoxid samt öka sensitiviteten av lösligt guanylatcyklas för kvävemonoxid. Macitentan och riociguat finns nu tillgängliga med läkemedelssubvention. Det finns dessutom ytterligare nya läkemedel på väg. När det gäller prostacyklinanaloger har effekterna hittills varit dramatiska vid systemisk tillförsel medan perorala formuleringar har varit behäftade med låg biotillgänglighet och blygsam verkningsgrad. Selexipag är en ny peroral prostanoidreceptoragonist med lovande korttidsresultat vad gäller hemodynamik och gångsträcka [20]. Utöver de tre grundläggande behandlingsspåren pågår försök med tyrosinkinashämmare som imatinib (PDGF-mekanism) och serotoninhämmare som tergurid [15].
För en del patienter där farmakologisk behandling inte varit framgångsrik kan utredning för lungtransplantation bli aktuell.
Sammanfattning
Sammanfattningsvis är sjukdomen pulmonell arteriell hypertension fortsatt allvarlig men framsteg har nåtts gällande systematisering av såväl definitioner som behandlingsprinciper. Samarbetet i Sverige genom Svensk förening för pulmonell hypertension är ytterst värdefullt både kliniskt och forskningsmässigt. Det nationella kvalitetsregistret SPAHR möjliggör jämförelser och utgör en god grund för lokala förbättringsarbeten samt forskning och utveckling genom samarbete både nationellt och internationellt.
Potentiella bindningar eller jävsförhållanden: Kjell Jansson har varit primary eller co-investigator i PAH-studier för Glaxo SmithKline, Actelion Pharmaceuticals, Pfizer och Bayer Health Care samt i kliniska hjärttransplantations-/immunsuppressionsstudier för Novartis. Stefan Söderberg är och har varit primary eller co-investigator i PAH-studier för Glaxo SmithKline, Actelion Pharmaceuticals, Pfizer, Novartis och Bayer Health Care samt varit engagerad i rådgivande kommittéer för Actelion Pharmaceuticals, Bayer, Eli-Lilly och Sanofi.Flemming Larsen har varit engagerad i rådgivande kommittéer för Actelion Pharmaceuticals och för Schering AG (Bayer HealthCare). Göran Rådegran är och har varit primary eller co-investigator i PAH-studier för Glaxo SmithKline, Actelion Pharmaceuticals, Pfizer, Bayer Health Care och United Therapeutics samt i kliniska hjärt-transplantations-/immunsuppressionsstudier för Novartis. Vidare har Rådegran varit engagerad i rådgivande kommittéer för Actelion Pharmaceuticals, Bayer, Eli-Lilly och Sanofi.
Fakta 1. Klassifikation av pulmonell hypertension
1. Pulmonell arteriell hypertension
1’. Pulmonell veno-ocklusiv sjukdom och/eller kapillär
hemangiomatosis
1’’. Persisterande pulmonell hypertension hos nyfödda
Undergrupper i grupp 1:
- Idiopatisk pulmonell arteriell hypertension
Hereditär eller familjär pulmonell arteriell hypertension
Pulmonell arteriell hypertension orsakad av läkemedel och gifter
Associerad pulmonell arteriell hypertension vid
− Bindvävssjukdom
− HIV-infektion
− Portal hypertension
− Medfödda hjärtfel
− Schistosomiasis
2. Pulmonell hypertension vid vänstersidig hjärtsjukdom
3. Pulmonell hypertension vid lungsjukdom och/eller hypoxemi
4. Pulmonell hypertension vid kronisk tromboemboli
5. Pulmonell hypertension av annan orsak
Referenser
- Ekmehag B. Pulmonell arteriell hypertension. Nya behandlingsprinciper ger förbättrad behandling vid ett ovanligt tillstånd. Läkartidningen. 2009;34:2057-61. http://lakartidningen.se/Functions/OldArticleView.aspx?articleId=12438
- D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343-9.
- Svenska pulmonell arteriell hypertension-registret (SPAHR). Årsrapport 2014. Uppsala: Svensk förening för pulmonell hypertension; 2015.
- Hoeper M, Bogaard H, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 -Suppl):-D42-50.
- Simonneau G, Gatzoulis M, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):–D34-41.
- Farber HW, Loscalzo J. Mechanism of disease: pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655-65.
- Schermuly RT, Ghofrani HA, Wilkins MR, et al. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443-55.
- Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. http://eurheartj.oxfordjournals.org/content/early/2015/08/28/eurheartj.ehv317
- Nationella riktlinjer för hjärtsjukvård. Stöd för styrning och ledning. Remissversion. Stockholm: Socialstyrelsen; 2015. Artikelnr 2015-1-12.
- Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France. Results from a national registry. Am J Respir Crit Care Med. 2006;173:1023-30.
- Barst RJ, Chung L, Zamanian RT, et al. Functional class improvement and 3-year survival outcomes in patients with pulmonary arterial hypertension in the REVEAL Registry. Chest. 2013;144(1):160-8.
- Sitbon O, Morell NW. Pathways in pulmonary arterial hypertension. Eur Resp Rev. 2012;21(126):321-7.
- Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425-36.
- Tuder RM, Archer SL, Dorfmüller P, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D4-12.
- Gomberg-Maitland M, Bull TM, Saggar T, et al. New trial designs and potential therapies for pulmonary artery hypertension. J Am Coll Cardiol. 2013;62(25 -Suppl):D82-91.
- Kylhammar D, Persson L, Hesselstrand R, et al. Prognosis and response to first-line single and combination therapy in pulmonary arterial hypertension. Scand Cardiovasc J. 2014;48(4):223-33.
- Galiè N, Barberà JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. New Eng J Med. 2015;373:834-44.
- Pulido T, Adzerikho I, Channick RN, et al; SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809-18.
- Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary hypertension. N Engl J Med. 2013;369:330-40.
- Simonneau G, Torbicki A, Hoerper AA, et al. Selexipag; an oral, selective prostacycline receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J 2012;40:874-80.