Sammanfattat
Prevalensen av homozygot alfa-1-antitrypsinbrist (AAT-brist) (fenotyp PiZZ) är 1/1 600 i Europa. AAT-brist är den vanligaste orsaken till neonatal hepatit och kolestatisk leversjukdom hos barn.En tredjedel utvecklar levercirros i vuxen ålder.
Leverförfettning är vanlig vid cystisk fibros. Cirka 5 procent utvecklar mer avancerad leversjukdom. Symtom på portal hypertension uppträder tidigare än symtom på leversvikt.
Wilsons sjukdom ska misstänkas hos unga patienter med akut leversvikt i kombination med hemolytisk anemi. Hos unga vuxna med parkinsonliknande sjukdomsbild och leverpåverkan ska Wilsons sjukdom uteslutas.
Glykogeninlagringssjukdomarna – främst von Gierkes sjukdom – ska misstänkas hos spädbarn med återkommande hypoglykemier och leverförstoring. Hos minst 50 procent av de patienter som når vuxen ålder utvecklas leveradenom med maligniseringspotential.
Flera ovanliga metabola sjukdomar kan kliniskt presentera sig som bl a leversjukdom. Förutom de sjukdomar som beskrivs i denna artikel – alfa-1-antitrypsinbrist, cystisk fibros, Wilsons sjukdom och glykogeninlagringssjukdomarna – hör hepatiska porfyrier till gruppen.
Leversjukdom vid alfa-1-antitrypsinbrist
Alfa-1-antitrypsinbrist är en relativt vanlig autosomal sjukdom, som alltid bör inkluderas i differentialdiagnostiska överväganden hos patienter med oklar transaminasstegring eller kronisk hepatit, levercirros och hepatocellulär cancer av oklar etiologi [1]. Alfa-1-antitrypsin (AAT), en alfa-1-proteasinhibitor, är det kvantitativt dominerande serinproteinaset i plasma. Flera välkarakteriserade mutationer i AAT-genen på kromosom 14 förorsakar nedsatt syntes och sänkta plasmakoncentrationer av denna viktiga proteinasinhibitor (Pi). Bland vuxna europeér är 1/40 bärare av Z-brist-allelen, medan 1/1 600 har homozygot brist (PiZZ). Kliniskt viktigast är PiZZ-bristen med 15 procent av normal plasmakoncentration och PiSZ med 37 procent [2] (Tabell I).
Den viktigaste biologiska uppgiften för alfa-1-antitrypsin är att neutralisera granulocytelastas, en funktion som hämmas av rökning. Otillräcklig hämning av detta enzym innebär risk för degradering av elastiska fibrer, främst i lungorna. Alfa-1-antitrypsinbrist misstänks vid låga nivåer av proteinet vid plasmaproteinanalys eller svagt AAT-band vid plasmaelektrofores. Pi-typen fastställs med hjälp av isoelektrisk fokusering eller genotypning.
Alfa-1-antitrypsin syntetiseras i levern, vilket påverkas av nedsatt leverfunktion. Koncentrationen stiger relativt snabbt vid inflammatoriska tillstånd, även vid heterozygota genetiska defekter, och alfa-1-antitrypsin räknas därför till kategorin akutfasproteiner. Det är således viktigt att värdera plasmanivån i förhållande till andra akutfasproteiner. Nivån kan särskilt väl användas som markör för aktiviteten av inflammation i levern. Koncentrationen ökar även under graviditet och vid användning av östrogenhaltiga p-piller [3].
Mutationen (aminosyrautbyte) i Z-proteinet gör att proteinet tenderar att polymerisera redan i hepatocyternas endoplasmatiska retikel i stället för först i samband med inaktivering av elastas. Polymeriseringen av det defekta proteinet leder till att det inte utsöndras på normalt sätt från hepatocyterna utan i stället orsakar apoptos eller nekros av hepatocyterna med fibrosbildning som följd. I mikroskopet syns det polymeriserade antitrypsinet som inklusionskroppar i hepatocyterna. PiS-proteinet aggregerar inte på samma sätt, utan har endast en fördröjd utsöndring och leder därför inte till cellskada i levern. Den sällsynta Pi0-fenotypen med avsaknad av alfa-1-antitrypsin i plasma är inte förenad med leversjukdom.
Den heterozygota PiZ-formen anses öka risken för leversjukdom något, speciellt i kombination med något annat etiologiskt agens, och är dessutom överrepresenterad vid kryptogen leversjukdom.
Klinisk presentation. Alfa-1-antitrypsinbrist är den vanligaste orsaken till neonatal hepatit och kolestatisk leversjukdom hos barn. Cirka 40 procent av dessa barn får kvarstående leversjukdom, varav en mindre andel progredierar till cirros med portal hypertension i tidig vuxenålder [4]. Hos 10–20 procent av individerna med PiZZ debuterar leversjukdom i stället i vuxen ålder, oftast i övre medelåldern hos icke-rökande individer som inte utvecklat lungsjukdom [3]. Leversjukdomen progredierar långsamt och kan leda till cirros med klart ökad risk för hepatocellulär cancer, leversvikt och portal hypertension. En obduktionsstudie från Malmö talar för att cirka en tredjedel av PiZZ-patienterna utvecklar levercirros, men att PiZZ inte sällan förlöper symtomfritt utan att diagnostiseras.
Behandling. Det finns ingen farmakologisk behandling av alfa-1-antitrypsinbrist med avseende på leversjukdom. Substitutionsbehandling har således ingen effekt på leversjukdomen. Det är viktigt att undvika ytterligare belastning på levern. Alkoholintaget bör därför begränsas och vaccination mot hepatit B-virus övervägas. Vid terminal leversjukdom är levertransplantation en botande behandling mot levercirrosen och återställer dessutom normal produktion av alfa-1-antitrypsin. Patienter med levercirros betingad av alfa-1-antitrypsinbrist bör inkluderas i övervakningsprogram avseende hepatocellulär cancer.
Leversjukdom vid cystisk fibros
Cystisk fibros är en av de vanligaste ärftliga sjukdomarna och drabbar 1/4 500–6 500 nyfödda i Sverige, dvs ca 14–20 barn per år. Sjukdomen är autosomalt recessivt ärftlig och orsakas av en mutation i genen som kodar för CFTR (cystic fibrosis transmembrane conductance regulator), ett epitelialt protein som transporterar klorid och bikarbonat. Sjukdomen drabbar främst lungor, pankreas, tarm, svettkörtlar och lever samt hos män funikeln. Dödligheten var tidigare hög i främst lungsjukdomen, men med modern behandling med energität nutrition, andningsgymnastik, mukolytika och antibiotika har medellivslängden kraftigt förbättrats och är i dag ca 50 år.
I takt med förlängd medellivslängd diagnostiseras allt fler patienter med cystisk fibros och leversjukdom relaterad till grundsjukdomen. Vanligast är leverförfettning, som ses hos upp till 60 procent av patienterna. Leverförfettningen kan kopplas dels till malnutrition, dels till mutationer associerade med lipidrubbning som ger brist på vissa essentiella fettsyror [5]. Cirka 25 procent av patienterna har förhöjda levervärden, men leversjukdom annan än leverförfettning diagnostiseras hos endast ca 5 procent, oftast före 10 års ålder. CFTR-proteinet uttrycks även i gallvägsepitelceller [6]. Vid cystisk fibros skadas gallgångarna, och inflammation och fibros i och kring dessa leder till fokal gallgångsfibros med förändringar som liknar skleroserande kolangit. Förändringarna kan progrediera och multilobulär biliär cirros utvecklas.
Klinisk presentation. Symtom på leversjukdom saknas oftast innan cirroskomplikationer med portal hypertension utvecklats. Leverförstoring med eller utan patologiska levervärden är det vanligaste kliniska fyndet och kan återspegla leverförfettningen. Fast, nodulerad leverförstoring, ofta begränsad till vänsterloben, är tillsammans med mjältförstoring tecken på cirrosutveckling med portal hypertension. Symtom på leversvikt med ikterus, ascites, encefalopati och stigande INR uppträder senare än symtomen på portal hypertension.
Ultraljudsundersökning av levern säkerställer ofta diagnosen leverförfettning då mer än en tredjedel av levercellerna innehåller fett. Ultraljudsundersökning som visar ojämn leveryta, mjältförstoring och – vid komplettering med dopplerundersökning av kärlen – utveckling av portosystemiska kollateraler säkerställer cirrosdiagnosen. Ultraljudsundersökning av levern kan dock visa normala fynd även vid betydande leverfibros [7].
Gallvägsförändringar liknande dem vid skleroserande kolangit kan påvisas hos hälften av patienterna med magnetresonanskolangiografi [8]. Transient elastografi (Fibroscan) som mäter leverstelheten noninvasivt är otillräckligt validerad vid leversjukdom vid cystisk fibros.
Leverbiopsi är den bästa metoden att bedöma graden av fibros i levern och risken för klinisk signifikant leversjukdom, men eftersom fibrosen ofta är fokal förordas två instick vid biopsitagning [9].
Behandling. Utöver sedvanlig behandling med hyperalimentation (150 procent av beräknat behov), andningsgymnastik, mukolytika, fettlösliga vitaminer och substitution med pankreasenzymer används ursodeoxicholsyra i dosen 15–20 mg/kg kroppsvikt för att stimulera gallflödet vid tecken på biokemisk leverpåverkan. Behandlingen förbättrar levervärdena, men effekt på överlevnad eller transplantationsbehov har inte kunnat visas [10, 11]. Behandlingen av esofagusvaricer följer rekommendationerna [12]. Patienter med cystisk fibros tål dock betablockad sämre än andra på grund av lungsjukdomen, vilket innebär att bandligering av varicer och transjugulär intrahepatisk portosystemisk shunt (TIPS) kan få högre prioritet som primär- och sekundärprofylax mot blödning från esofagusvaricer vid cystisk fibros.
Levertransplantation utförs hos ett fåtal patienter med progredierande leversvikt eller intraktabel varixblödning och relativt välbevarad lungfunktion. Femårsöverlevnaden efter levertransplantation är ca 80 procent [13, 14]. Kombinerad lever- och lungtransplantation har sämre prognos.
Leversjukdom vid Wilsons sjukdom
Wilsons sjukdom är en sällsynt recessivt ärftlig sjukdom med en incidens som beräknats till 1/30 000, dvs det föds 3–4 individer per år i Sverige med Wilsons sjukdom [15]. Sjukdomen orsakas av ett hundratal mutationer i en gen (ATP7B på kromosom 13) som kodar för ett koppartransporterande ATPas. Proteinet är lokaliserat i det kanalikulära membranet i hepatocyten. Vid Wilsons sjukdom är ATPas defekt, vilket medför att utsöndringen av koppar i gallan är nedsatt, och koppar ansamlas i stället i främst lever, hjärna och kornea, där en s k Kayser–Fleischer-ring kan påvisas med spaltlampsundersökning (Figur 1).
Klinisk presentation. Kliniskt debuterar sjukdomen oftast mellan 5 och 35 års ålder. Wilsons sjukdom presenterar sig på olika sätt med antingen symtom på leversjukdom eller neurologiska eller psykiatriska symtom [16]. Asymtomatiska fall finns och upptäcks oftast vid släktundersökning. Cirka en tredjedel debuterar med leversymtom. Akut leversvikt med hemolytisk anemi och subnormalt ALP-värde och bara lätt förhöjda transaminaser hos en ung patient ska väcka misstanke om Wilsons sjukdom. Bara hälften har Kayser–Fleischerring i kornea.
Trots den akuta debuten har merparten redan utvecklat cirros i detta skede. Obehandlad är dödligheten nästan 95 procent, och levertransplantation ska övervägas. Sjukdomsdebuten kan vara mer smygande och omöjlig att kliniskt skilja från annan kronisk leversjukdom som kronisk hepatit och kompenserad eller dekompenserad cirros.
Neurologiska symtom på Wilsons sjukdom debuterar ofta i ung vuxen ålder. Symtomen kan initialt vara diskreta och intermittenta, men de kan progrediera snabbt. De består främst av tremor, akinesi/rigiditet och dysartri samt ataxi, dystoni och terminalt ibland svår kognitiv svikt. Även psykiatriska symtom med personlighetsstörning och psykotiska symtom som paranoia och depression kan uppträda. Merparten patienter med neurologiska symtom har Kayser–Fleischer-ring, och hälften har avancerad fibros eller cirros i levern. Wilsons sjukdom ska uteslutas hos unga vuxna med nytillkomna neurologiska symtom och leverpåverkan.
Diagnosen Wilsons sjukdom vilar på en sammanvägd bild av klinik, påvisning av Kayser–Fleischer-ring, lågt S-ceruloplasmin och hög kopparutsöndring i urinen. Diagnosen kan bekräftas med gentest (på Akademiska sjukhuset i Uppsala efter kontakt) eller mätning av kopparinnehåll i levervävnad. En europeisk diagnostisk algoritm finns [17].
Behandling. Obehandlad är sjukdomen dödlig främst på grund av leversvikt, mer sällan på grund av progredierande neurologisk sjukdom. Behandlingen är livslång och består av läkemedel som via kelatbindning av koppar ökar urinutsöndringen av överskottet (D-penicillamin, trientin, ammoniumtetratiomolybdat) eller zink som hämmar upptaget av koppar från tarmen. Efter initial mobilisering av kopparöverskottet kan de flesta patienter underhållsbehandlas med zinktillskott.
Med adekvat och tidig behandling av Wilson-patienter utan cirros eller med kompenserad cirros är prognosen troligen god. Vid akut leversvikt är transplantation oftast livräddande. Neurologiska symtom förefaller mindre reversibla trots adekvat behandling. Släktundersökning är viktig, eftersom risken för att ett syskon har mutationen och därmed risk att utveckla klinisk sjukdom är 25 procent.
Glykogeninlagringssjukdomar
Glykogeninlagringssjukdomar är en grupp ärftliga sjukdomar som orsakas av onormal omsättning av glykogen, vilket leder till onormal inlagring av glykogen i bl a levern. Den vanligaste – von Gierkes sjukdom – finns hos ca 1/100 000 nyfödda, dvs 1 barn per år i Sverige. Sjukdomen ärvs autosomalt recessivt och leder till brist på enzymet glukos-6-fosfatas, som katalyserar nedbrytningen av glykogen till glukos i levern.
Klinisk presentation. Sjukdomen diagnostiseras oftast under första levnadsåret, och kliniskt karakteriseras bilden av förstorad lever och återkommande hypoglykemier [18]. Enzymbristen leder till andra metabola rubbningar som mjölksyraacidos, hyperlipemi och hyperurikemi. Vid tidig diagnos och med adekvat behandling uppnår patienterna vuxen ålder, men tillväxthämning är vanlig och på sikt kan patienterna utveckla nefropati, hypertoni, inflammatorisk tarmsjukdom och osteopeni. Leveradenom ses hos minst hälften av patienterna vid 20 års ålder [19], och hos ca 10 procent blir de maligna [20]. Diagnosen säkerställs genom genetisk analys.
Behandling. Behandlingen syftar till att behålla fysiologiska blodglukosnivåer och består av diet med majsstärkelse. På grund av den höga förekomsten av leveradenom bör patienterna följas med ultraljudskontroller av levern och levertumörer behandlas. Levertransplantation har genomförts som behandling hos patienter med grav metabol sjukdom samt vid levertumörer och leversvikt. Levertransplantation botar den metabola rubbningen.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
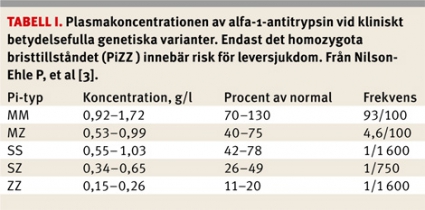
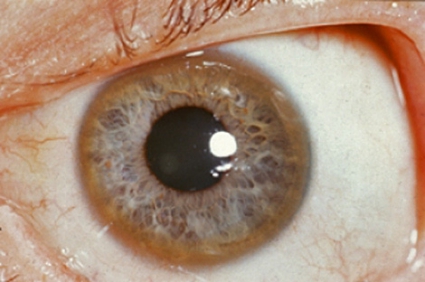
Figur 1. Kayser–Fleischer-ring hos patient med Wilsons sjukdom. Inlagring av brunt kopparhaltigt pigment i korneas periferi.
Referenser
1. Eriksson S, Carlson J, Velez R. Risk of cirrosis and primary liver cancer in alpha-1-antitrypsin deficiency. N Engl J Med. 1986;314:736-9.
2. Fairbanks KD, Tavill AS. Liver disease in alpha-1-antitrypsin deficiency : A review. Am J Gastroenterol. 2008;103:2136-41.
3. Nilsson-Ehle P, Berggren Söderlund M, Theodorsson E. Laurells klinisk kemi i praktisk medicin. Lund: Studentlitteratur: 2012. p. 123-4.
4. Sveger T. Liver disease in alpha-1-antitrypsin deficiency detected by screening of 200 000 infants. N Engl J Med. 1976;294:1316-21.
5. Lindblad A, Glaumann H, Strandvik B. Natural history of liver disease in cystic fibrosis. Hepatology. 1999;30:1151-8.
6. Kinnman N, Lindblad A, Housset C, et al. Expression of cystic fibrosis transmembrane conductance regulator in liver tissue from patients with cystic fibrosis. Hepatology. 2000;32:334-40.
7. Mueller-Abt PR, Frawley KJ, Greer RM, et al. Comparison of ultrasound and biopsy findings in children with cystic fibrosis related liver disease. J Cyst Fibros. 2008;7:215-21.
8. Durieu I, Pellet O, Simonot L, et al. Sclerosing cholangitis in adults with cystic fibrosis: a magnetic resonance cholangiographic prospective study. J Hepatol. 1999;30:1052-6.
9. Lewindon PJ, Shepherd RW, Walsh MJ, et al. Importance of hepatic fibrosis in cystic fibrosis and predictive value of liver biopsiy. Hepatology. 2011;53:193-201.
10. Colombo C, Battezzati PM, Podda M, et al. Ursodeoxycholic acid for liver disease associated with cystic fibrosis. A double-blind multicenter trial. Hepatology. 1996;23:1484-90.
11. Cheng K, Ashby D, Smyth R. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev. 2010;10:1-29.
12. Stål P, Stotzer PO, Söderlund C, et al. Handläggning av varicer i esofagus och ventrikel. Nationella riktlinjer 2009. Stockholm: Svensk gastroenterologisk förening; 2010.
13. Melzi ML, Kelly D, Colomco C, et al. Liver transplant in cystic fibrosis: a poll among European centres. A study from the European Liver Transplant Registry. Transpl Int. 2006;19:726-31.
14. Mendizabal M, Reddy KR, Cassuto J, et al. Liver transplantation in patients with cystic fibrosis. Analysis of united network for organ sharing data. Liver Transpl. 2011;17:243-50.
15. Reilly M, Daly L, Hutchinson M. An epidemiologogical study of Wilson’s disease in the Republic of Ireland. J Neurol Neurosurg Psychiatry. 1993; 56:298-300.
16. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012;56:671-85.
17. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson’s disease. Liver Int. 2003;23:139-42.
18. Rake JP, Visser G, Labrune P, et al. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of an European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002;161 Suppl 1:S20-34.
19. Talente GM, Coleman RA, Inoue F, et al. Glycogen storage disease in adults. Ann Intern Med. 1994;120:218-26.
20. Bianchi L. Glycogen storage disease I and hepatocellular tumours. Eur J Pediatr. 1993;152 Suppl 1:S63-70.