Sammanfattat
Ny populationsgenetik har identifierat ett samband mellan tarmens bakterieflora och en dysfunktionell epitelvägg som en viktig förklaring till det kroniska inflammatoriska tillståndet vid inflammatorisk tarmsjukdom (IBD). Den mest karakteriserade riskgenen hitintills är NOD2.
I bakteriefri miljö uppkommer ingen inflammation i djurmodeller för kolit. Antibiotika och probiotika påverkar sjukdomsförloppet hos patienter med IBD.
Tarmslemhinnans barriärfunktion är förändrad vid IBD och rubbningar i specialiserade epitelceller som Paneth-celler och follikelassocierat epitel kan ha betydelse för sjukdomsuppkomsten.
Dessa fynd ger stöd för att de immunologiska förändringarna vid IBD sannolikt beror på genetiska störningar i den medfödda immuniteten.
Kroniska inflammatoriska tillstånd i olika organsystem är vanliga och utmanande sjukdomar för dagens sjukvård. Sjukdomarna är till stor del polygenetiska och uppvisar symtombilder som sammantaget ger kliniskt relativt välgrundade diagnoser, t ex psoriasis, systemisk lupus erythematosus (SLE), multipel skleros (MS) och inflammatorisk tarmsjukdom (IBD). Dagens läroböcker beskriver ofta symtomen som organbundna företeelser intimt sammanlänkade med den aktuella inflammationshärden men diskuterar sällan de systembiologiska effekter som otvetydigt finns vid kroniska inflammationer som IBD (Figur 1).
Med hjälp av nya, kraftfulla populationsgenetiska metoder och utvecklandet av bra experimentella modellsystem har ett antal genetiska markörer identifierats, av vilka flera är gemensamma för de komplexa inflammatoriska sjukdomarna. Dessa fynd stödjer tanken om överlappande patofysiologiska signalvägar och hittills icke-identiferade samband mellan många olika kroniska inflammationssjukdomar och har lett till ökad kunskap kring uppkomstmekanismerna för kronisk inflammation och, inte minst, öppnat för nya angreppsvinklar för terapi.
Det klassiska »autoimmuna« synsättet (ibland nästan en »slaskdiagnos«) har gett oss föreställningen att störningar i vårt adaptiva immunsystem är orsaken till IBD. De nya fynden som kommit under senare år, framför allt genom identifiering av predispositionsgener, pekar i stället på att B- och T-lymfocyterna blir instruerade på ett felaktigt sätt av celler och signalsystem ingående i den medfödda immuniteten. Vi vill med denna översiktartikel ge en aktuell bild av forskningsfronten inom IBD och införliva nya potentiella spelare som är inblandade i sjukdomsprocessen.
Genetiska faktorer
I de numera klassiska tvillingstudier som gjordes av en forskargrupp i Örebro på 1980-talet [1] pekade man på att genetiskt predisponerande faktorer spelar en större roll för uppkomsten av Crohns sjukdom än för uppkomsten av ulcerös kolit. Tillgången till en karta av det mänskliga genomet öppnade sedan för en kartläggning av riskområden inom arvsmassan, och populationsgenetiska studier kunde tydligare avgränsa och identifiera riskområden på olika kromosomer som innehöll ett ökat antal genpolymorfier (naturligt förekommande genvarianter) hos patienter med Crohns sjukdom. Dessa områden benämndes IBD1 [2], IBD2 [3], IBD3 etc.
Det var därför inte förvånande att två forskargrupper år 2001 kunde rapportera identifieringen av genförändringen bakom IBD1, dvs NOD2 (även kallad CARD15) [4, 5]. NOD2-proteinet är en intracellulär receptor för delar av bakteriens cellvägg (muramyldipeptid) och förekommer framför allt i epitelceller och monocyter. Dessa studier pekade på att den symtombild som uppträder vid Crohns sjukdom kunde länkas samman med en signalväg som kan regleras genom den normala tarmfloran. Nya rön har identifierat många andra genlokus som predisponerar för Crohns sjukdom [6], och flertalet stödjer ett samband med tarmflorans bakterier och störningar i tarmens barriärfunktion [7]. Viktiga exempel är generna ATG16L1 och IRGM, som kodar för reglering av cellulär autofagi [8-10], som antas vara kopplad till försvaret mot intracellulärt växande bakterier.
Ytterligare stöd för överlappande signalvägar
Andra exempel är olika jonkanaler som IBD5 (OCTN1 och OCTN2), som är involverad i epitelcellernas membranfunktion [11], och kromosom 5p13.1, sannolikt i den del som kodar för prostaglandinreceptorn EP4 [12]. Nyligen har vi också identifierat en G-kopplad receptor, GPRA [13], som uppträder vid både ulcerös kolit och Crohns sjukdom och som tidigare rapporterats som en markör för astma [14]. Likaså har förändringar i generna för den s k NALP3-inflammasomen, ett proteinkomplex som är involverat i IL-1β-svaret på bakterietoxiner, betydelse för sjukdomsrisken vid Crohns sjukdom [15] (även i en svensk population [16]) och vid reumatoid artrit [17]. Samtliga dessa genetiska förändringar i dessa markörgener kan indirekt kopplas till störningar i barriäregenskaper och epitelväggsfunktion (Figur 2).
I en annan nyligen rapporterad studie från Cho och medarbetare fann forskargruppen att IL-23-receptorn är en genetisk riskfaktor hos patienter med Crohns sjukdom [18]. Det faktum att denna IL-23-receptor också uppträder som riskfaktor vid psoriasis ger ytterligare stöd för möjliga överlappande signalvägar hos olika kroniska inflammationssjukdomar med polygenetiskt ursprung.
Genetiska varianter ger dock inte hela förklaringen till IBD-sjukdomarna. I olika populationer har man visat att den starkaste genetiska faktorn, dvs polymorfier i NOD2, kraftigt (ca 30 gånger) ökar risken för att insjukna i Crohns sjukdom. Men de flesta personer som har NOD2-förändringar blir inte tarmsjuka. NOD2-polymorfier kan inte förklara mer än högst en tredjedel av crohnfallen i Europa. I de nordiska länderna är polymorfier i NOD2 relativt ovanliga [19], ändå är incidensen och prevalensen av Crohns sjukdom minst lika hög som i Nordamerika och Storbritannien. I asiatiska populationer, där incidensen nu galopperar, förekommer polymorfierna knappast alls [20, 21]. Detta talar för att det utöver en genetisk predisposition behövs andra faktorer som utlöser och/eller driver inflammationen. En uppenbar inflammationsdrivande faktor vid IBD är tarmens egen bakterieflora.
Tarmflorans roll
Vi vet väldigt lite om den bomb av bakterier (1 kg) som finns i vår mag–tarmkanal; t ex kan de flesta av dessa ca 400–500 olika stammar inte odlas. Vi befinner oss alltså ännu i tidigt skede av den cellulära mikrobiologin, men de ca 50-talet djurmodellerna för IBD (t ex MDR1, N-cadherin, PPARγ och konditionell knockout av IL-2, IL-12, IL-10, NFκB) ger en entydig bild: om dessa djur vistas under bakteriefria förhållanden (gnotobiotisk miljö) förekommer inte inflammation i mag–tarmkanalen [7]. Ofta krävs bara tillförsel av en enstaka icke-patogen bakteriestam för att utlösa tarminflammation [22], vilket gör det uppenbart att interaktionen mellan lumenfaktorer (bakterier) och värdfaktorer (epitel och kroppens försvarsmekanismer) är viktiga för inflammationen. Andra studier ger vid handen att bakteriefloran hos IBD-patienter i ökad omfattning innehåller bakteriestammar som fäster i mukuslagret alldeles intill epitelet.
I både ileum [23] och kolon [24] har särskilda stammar av E coli som är adherenta till epitelcellerna identifierats. Dessa E coli har också egenskaper som gör att de kan ta sig in via epitelcellerna [23, 25] och överleva i makrofager i slemhinnan [24, 26], vilket gör att de kan underhålla en kronisk inflammation. Mer oklart är ifall dessa stammar är en konsekvens av en dysfunktionell barriär eller om de aktivt kan initiera den inflammatoriska processen i samband med allvarlig stress, t ex infektion, stort trauma eller långvarig psykologisk stress. Kort sagt kan det vara så att tarmfloran hos IBD-patienter genom sin komposition ger ett sämre skydd än den hos människor som inte har IBD.
Experimentella data [27] och konfirmerande fynd från en studie på svenska tvillingar [28] som talar för en sådan skillnad i bakteriefloran har nyligen rapporterats. Detta område är för närvarande mycket forskningsintensivt, och detaljerade och omfattande studier både i djurmodeller och hos människa pågår för fullt. Kliniska observationer ger också stöd för bakteriernas betydelse vid Crohns sjukdom. Inflammationen uppstår oftast i tarmregioner där bakteriemängden är hög (ileum–kolon) och avlastning av lumeninnehållet med t ex ileostomi kan läka ut inflammationen [29]. Dessutom återkommer inflammation i ileum nästan omgående efter att tarmkontinuiteten återställts [30].
Antibiotikabehandling har positiva effekter, åtminstone i undergrupper av patienter med crohn [31]. När det gäller probiotiska effekter är bilden betydligt mer komplex, och någon klar bild av att probiotikabehandling faktiskt kan hjälpa IBD-patienter finns inte, även om enstaka studier rapporterat positiva resultat med bakteriestammen Nissle [32, 33]. Ett positivt undantag finns: man har funnit att probiotikablandningen VSL#3 är den mest effektiva underhållsbehandlingen vid kronisk pouchit efter bäckenreservoarkirurgi för ulcerös kolit [34], och detta är nu rekommenderad behandling för denna patientgrupp [35].
Barriärfunktionen
Samtidigt som tarmslemhinnans celler ska absorbera näringsämnen och vätska är de också kroppens viktigaste barriärer mot yttre miljöpåverkan och utsätts oavbrutet för ämnen som kan påverka dess funktion: bakterier, miljögifter, fria syreradikaler och höga koncentrationer av näringsämnen vid onormalt födointag är några exempel. Tarmbarriären består av flera olika komponenter: en fysisk diffusionsbarriär, en reglerad fysiologisk och enzymatisk barriär och en immunologisk barriär (Figur 2).
Ett kontinuerligt skikt av epitelceller, som sammanfogas av ett apikalt proteinkomplex, s k tight junctions, begränsar både paracellulärt och transcellulärt upptag och utgör därigenom huvudkomponenten i barriären. Vi vet t ex i dag att tarmfloran kan reglera gener som styr uppbyggnaden av tight junctions. En dynamisk, reglerad sekretion av vätska och IgA-innehållande mukus sköljer bort, späder och binder skadliga ämnen. Tarmens försvarsmekanismer inkluderar också propulsiv tarmmotorik, som motverkar bakteriell överväxt i lumen. Ett flertal studier har visat att uppbyggnaden av vår barriär till normal funktion framför allt sker postembryonalt och är ett intimt samspel mellan våra epitelceller, vårt mukuslager, dess immunceller i lamina propria och vår egen tarmflora [36-38].
Det finns experimentella fynd som stödjer tanken om att en störd barriärfunktion har betydelse vid IBD. Vid ulcerös kolit finns indikationer på att barriärfunktionen i kolon är förändrad vid aktiv sjukdom vad gäller både paracellulär [39, 40] och transcellulär [41, 42] permeabilitet, och man tror att IL-13 kan vara en viktig signalsubstans i denna process [43]. Vidare har man observerat låga nivåer i epitelcellerna av PPARγ, en av de nukleära receptorer som kan inhibera NFκB [44]. Hos patienter med crohn har ökad genomsläpplighet i tarmen rapporterats, och man ser en störd intestinal permeabilitet hos en betydande andel av förstagradssläktingar till crohnpatienter [45, 46]. Dessa fynd har nyligen länkats till polymorfier i NOD2-genen [47].
Barriärstörningen vid Crohns sjukdom medieras till stor del via TNFα vad gäller både transcellulärt upptag av molekyler [48] och paracellulär permeabilitet [49]. Att läkemedlet infliximab motverkar den TNFα-inducerade apoptosen av epitelceller ger en möjlig förklaring till de ibland så dramatiskt positiva kliniska resultaten [50]. Man har också funnit förändringar i olika specifika epitelcellstyper vid Crohns sjukdom. Paneth-cellerna, som ses som granulerade celler i basen på kryptorna (framför allt i ileum), har som viktig uppgift att producera antibakteriella peptider, s k defensiner. Det finns både funktionella [51, 52] och genetiska [53] bevis för att produktion och sekretion av α-defensiner är förändrad vid Crohns sjukdom. I Paneth-cellerna är också NOD2-proteinet starkt uttryckt [54].
En tänkbar mekanism för hur muterat NOD2 är kopplat till symtombilden vid crohn är att störd avkänning av bakterierna leder till defekt sekretion av de antibakteriella peptiderna i ileum. Follikelassocierat epitel, som täcker lymfolliklarna i de peyerska placken, är funktionellt specialiserat på antigenupptag för att hjälpa immunsystemet att läsa av lumeninnehållet. Vi har visat att denna funktion är förändrad vid crohn, med ökat upptag av bakterier via epitelet till dendritiska celler, vilket i sin tur medför ett ökat proinflammatoriskt svar [55, 56]. Detta samspel mellan epitelet och kroppens försvarsmekanismer är ytterligare en viktig faktor som kan bidra till initiering eller underhåll av inflammation.
Cytokinernas roll
Olika cytokiner har rapporterats orsaka inflammationen vid IBD. Vid Crohns sjukdom har den förhärskande immunologiska hypotesen varit att inflammationen uppstår som en konsekvens av patologiska T-hjälparlymfocyter (Th1 CD4-positiva celler) riktade mot vår egen tarmflora och underhålls av cytokinen IL-12. Vid en närmare molekylär analys fann man dock att IL-23 (som delar ena subenheten (IL-12p40) med IL-12) är en betydligt viktigare regulator för mukosans immunitet. Liksom IL-12 produceras IL-23 av dendritiska celler, som konstant läser av vår normala tarmflora.
Vid intestinal inflammation fann man en lokal dysreglering av IL-23-receptorn i mukosan men normalt uttryck av IL-23 i mjälte och lever [57]. När så populationsgenetikerna visade att IL-23-receptorn kunde länkas som genetisk riskfaktor till crohn förändrades vårt synsätt, och man kan skönja ett paradigmskifte i synen på crohn och cytokindriven inflammation [18]. Proinflammatoriska cytokiner frisätts mer som en konsekvens av en dysreglerad barriärfunktion som leder till aktivering av IL-23-producerande dendritiska celler. IL-23 i sin tur reglerar en liten grupp av IL-17-producerande Th-celler (Th17) som styr och underhåller mukosans immunitet. Att selektivt angripa IL-23-receptorn och dess signaleringsväg framstår därför som en mer logisk väg att inhibera inflammation vid crohn än att använda systemisk immunsuppression [18, 57]. Detta återstår dock att bevisa i pågående kliniska behandlingsstudier.
Nukleär faktor kappa B
Den kroniska inflammationen i mag–tarmkanalen vid IBD underhålls av proinflammatoriska cytokiner som IL-1, IL-6 och TNFα, vilka bidrar till vävnadsdestruktion. En gemensam nämnare som reglerar aktiviteterna av dessa inflammationsmediatorer är transkriptionsfaktorn nukleär faktor kappa B (NFκB). Denna faktor identiferades 1986 och har sedan dess påvisats vara den kanske viktigaste regulatorn för mukosaimmunitet. NFκB är en DNA-bindande regulator som består av fem subenheter, p65 (RelA), c-Rel, RelB, p50 och p52, som kan homo- och heterodimerisera med varandra. NFκB är bundet till NFκB-inhibitorer (IκBα, IκBβ eller IκBε) som finns lokaliserade i cytoplasman. Dessa IκB-varianter kan också stänga av NFκB genom att binda till NFκB i cellkärnan och exportera NFκB till cytoplasman. Icke-stimulerade celler härbärgerar NFκB utanför cellkärnan.
Aktiveringen av NFκB kan medieras genom två vägar, den klassiska och den alternativa. Oavsett signalväg leder NFκB-aktivering till translokation av NFκB till cellkärnan och aktivering av målgener för NFκB. Den klassiska vägen utnyttjar bakteriella membrankomponenter (t ex lipopolysackarider), virus och DNA-skadande ämnen som aktiverar det s k Iκ-kinas(IKK)-komplexet, som i en serie fosforyleringar, framför allt genom subenheten IKKb, resulterar i proteindegradering av IκB som frigör NFκB att translokera till cellkärnan. Genom den alternativa aktiveringsvägen kan CD40-receptorn (medlem av TNF-receptorfamiljen) initiera en klyvning av ett prekursorprotein (p100) till det slutgiltiga aktiva proteinet (p52). Oberoende av vilken signalväg som aktiveras är de nedströms aktiverade generna intimt kopplade till inflammation och immunitet och till formering av stora proteinkomplex kallade »inflammasome protein complexes«.
Vi och andra har beskrivit att i princip alla läkemedel som i dag används för behandling av IBD (kortikosteroider, sulfasalazin, metorexat och anti-TNFα-antikroppar) kan stänga av NFκB-aktiviteten. Exempelvis aktiverar steroidbehandling utrrycket av IκBα, som därigenom kan exportera NFκB ur cellkärnan. Resultatet är att epitelceller, makrofager och endoteliala celler uppvisar lägre nivåer av NFκB hos behandlade patienter än hos obehandlade [58].
Även om NFκB framstår som ett attraktivt målprotein för behandling av en kronisk inflammation som IBD är det viktigt att komma ihåg att NFκB också är en viktig regulator av cellernas normala fysiologi. NFκB reglerar kroppens lymfocyter, är helt nödvändig för att skydda oss mot bakteriella infektioner, är en essentiell komponent i reparationen av skadad vävnad och är en av de viktigaste regulatorerna för normal levercellsfunktion. Nya terapivägar som syftar till att stänga av NFκB bör därför utvecklas på ett sätt som tillåter selektiv avstängning av NFκB lokalt i det inflammerade området.
Nya aktörer i den kroniska inflammationen
Nya experimentella rön ger stöd för att nukleära receptorer på flera sätt kan påverka den kroniska inflammationen vid IBD. Receptorer som PPARγ och PXR har nyligen visat sig vara viktiga regulatorer av barriärfunktionen i mag–tarmkanalen genom att hämma NFκB [59]. De sammankopplande länkarna mellan dessa receptorer är NFκB och överlappande signalvägar till kroppens detoxifieringssystem [Pettersson S et al, Stockholm, pers medd; 2009]. Den nukleära receptorn LRH-1, som bl a styr fettomsättning och celltillväxt, kan också inhibera kronisk inflammation genom att aktivera signalvägar som leder till utsöndring av glukokortikoider [60].
Konklusion
Sammanfattningsvis tyder mycket på att IBD uppkommer när en genetiskt predisponerad individ (med störningar i barriärfunktionen och/eller det medfödda immunsystemet) utsätts för utlösande omgivningsfaktorer, där vår normala tarmfloras komposition verkar vara viktig för att underhålla den kroniska inflammationen. De nya markörer som identifierats kommer att öka kunskapen kring vilka signalvägar och patofysiologiska processer som styr vid IBD, och intensifierad forskning inom dessa områden kommer att öppna nya vägar för läkemedelsutveckling och förbättra behandlingsmetoderna. Vidare kommer dagens pågående genomik- och proteomikforskning att kompletteras med en omfattande kartläggning av tarmfloran, s k mikrobiomik. Det är fascinerande att vara med i forskningsfältet kring patogenesen för IBD. Under de närmaste åren kommer nya intressanta rön att se dagens ljus som tveklöst väsentligt kommer att öka våra kunskaper kring uppkomsten av IBD.
*
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
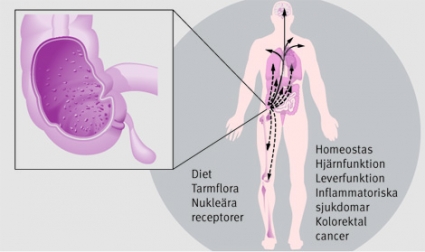
Figur 1. Inflammatorisk tarmsjukdom (IBD) är en systembiologisk inflammation. Kronisk inflammation (multipel skleros, astma, psoriasis, reumatoid artrit, IBD) i olika organsystem har visat sig delvis ha gemensamma genetiska störningar. Dessutom kan den lokala miljön i tarmen påverka hjärna, lever, leder, cancerutveckling etc. Denna nya kunskap har ändrat sättet att tänka kring uppkomsten av kroniska inflammationssjukdomar.
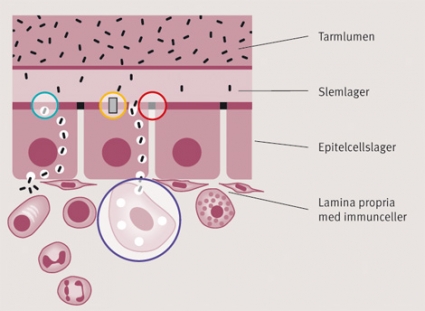
Figur 2. Tarmbarriären skyddar oss från tarmlumens enorma innehåll av antigener och mikroorganismer. De viktigaste komponenterna är slemlagret, epitelcellerna och det medfödda immunsystemet (innate immunity) i lamina propria. Cirklarna markerar cellulär lokalisation för några av de viktigaste genetiska förändringar som predisponerar för IBD. Blå cirkel = Bakterie–epitelinteraktion (NOD2, ATG16L1, NALP3). Gul cirkel = transportkanaler (OCTN1, OCTN2). Röd cirkel = Tight junction-området (DLG5). Lila cirkel = Dendritiska celler (IL-23, NALP3). Se texten för mer detaljer.
Referenser
(1) Tysk C, Lindberg E, Jarnerot G, Floderus-Myrhed B. Ulcerative colitis and Crohn's disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut 1988; 29(7):990-996.
(2) Hugot JP, Laurent-Puig P, Gower-Rousseau C, Olson JM, Lee JC, Beaugerie L et al. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature 1996; 379(6568):821-823.
(3) Satsangi J, Parkes M, Louis E, Hashimoto L, Kato N, Welsh K et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet 1996; 14(2):199-202.
(4) Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001; 411(6837):603-606.
(5) Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001; 411(6837):599-603.
(6) Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 2008; 40(8):955-962.
(7) Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007; 448(7152):427-434.
(8) Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007; 39(2):207-211.
(9) Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39(5):596-604.
(10) Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet 2007; 39(7):830-832.
(11) Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet 2004; 36(5):471-475.
(12) Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet 2007; 3(4):e58.
(13) D'Amato M, Bruce S, Bresso F, Zucchelli M, Ezer S, Pulkkinen V et al. Neuropeptide s receptor 1 gene polymorphism is associated with susceptibility to inflammatory bowel disease. Gastroenterology 2007; 133(3):808-817.
(14) Vendelin J, Pulkkinen V, Rehn M, Pirskanen A, Raisanen-Sokolowski A, Laitinen A et al. Characterization of GPRA, a novel G protein-coupled receptor related to asthma. Am J Respir Cell Mol Biol 2005; 33(3):262-270.
(15) Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat Genet 2009; 41(1):71-76.
(16) Schoultz I, Verma D, Halfvarsson J, Torkvist L, Fredrikson M, Sjoqvist U et al. Combined Polymorphisms in Genes Encoding the Inflammasome Components NALP3 and CARD8 Confer Susceptibility to Crohn's Disease in Swedish Men. Am J Gastroenterol 2009.
(17) Kastbom A, Verma D, Eriksson P, Skogh T, Wingren G, Soderkvist P. Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology (Oxford) 2008; 47(4):415-417.
(18) Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006; 314(5804):1461-1463.
(19) Torkvist L, Noble CL, Lordal M, Sjoqvist U, Lindforss U, Nimmo ER et al. Contribution of CARD15 variants in determining susceptibility to Crohn's disease in Sweden. Scand J Gastroenterol 2006; 41(6):700-705.
(20) Ouyang Q, Tandon R, Goh KL, Ooi CJ, Ogata H, Fiocchi C. The emergence of inflammatory bowel disease in the Asian Pacific region. Curr Opin Gastroenterol 2005; 21(4):408-413.
(21) Inoue N, Tamura K, Kinouchi Y, Fukuda Y, Takahashi S, Ogura Y et al. Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology 2002; 123(1):86-91.
(22) Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001; 291(5505):881-884.
(23) Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology 2004; 127(2):412-421.
(24) Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R et al. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology 2004; 127(1):80-93.
(25) Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest 2007; 117(6):1566-1574.
(26) Mpofu CM, Campbell BJ, Subramanian S, Marshall-Clarke S, Hart CA, Cross A et al. Microbial mannan inhibits bacterial killing by macrophages: a possible pathogenic mechanism for Crohn's disease. Gastroenterology 2007; 133(5):1487-1498.
(27) Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008; 105(43):16731-16736.
(28) Willing B, Halfvarson J, Dicksved J, Rosenquist M, Jarnerot G, Engstrand L et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn's disease. Inflamm Bowel Dis 2009; 15(5):653-660.
(29) Rutgeerts P, Goboes K, Peeters M, Hiele M, Penninckx F, Aerts R et al. Effect of faecal stream diversion on recurrence of Crohn's disease in the neoterminal ileum. Lancet 1991; 338(8770):771-774.
(30) D'Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn's disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology 1998; 114(2):262-267.
(31) Rutgeerts P, Van Assche G, Vermeire S, D'Haens G, Baert F, Noman M et al. Ornidazole for prophylaxis of postoperative Crohn's disease recurrence: a randomized, double-blind, placebo-controlled trial. Gastroenterology 2005; 128(4):856-861.
(32) Rembacken BJ, Snelling AM, Hawkey PM, Chalmers DM, Axon AT. Non-pathogenic Escherichia coli versus mesalazine for the treatment of ulcerative colitis: a randomised trial. Lancet 1999; 354(9179):635-639.
(33) Kruis W, Fric P, Pokrotnieks J, Lukas M, Fixa B, Kascak M et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 2004; 53(11):1617-1623.
(34) Gionchetti P, Rizzello F, Venturi A, Brigidi P, Matteuzzi D, Bazzocchi G et al. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119(2):305-309.
(35) Biancone L, Michetti P, Travis SP, Escher JC, Moser G, Forbes A et al. European evidence-based Consensus on the management of ulcerative colitis: Special situations. Journal of Crohn's and Colitis 2008; 2(1):63-92.
(36) Are A, Aronsson L, Wang S, Greicius G, Lee YK, Gustafsson JA et al. Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proc Natl Acad Sci U S A 2008; 105(6):1943-1948.
(37) Lundin A, Bok CM, Aronsson L, Bjorkholm B, Gustafsson JA, Pott S et al. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol 2008; 10(5):1093-1103.
(38) Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001; 291(5505):881-884.
(39) Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology 1999; 116(2):301-309.
(40) Almer S, Franzen L, Olaison G, Smedh K, Strom M. Increased absorption of polyethylene glycol 600 deposited in the colon in active ulcerative colitis. Gut 1993; 34(4):509-513.
(41) Schurmann G, Bruwer M, Klotz A, Schmid KW, Senninger N, Zimmer KP. Transepithelial transport processes at the intestinal mucosa in inflammatory bowel disease. Int J Colorectal Dis 1999; 14(1):41-46.
(42) Nejdfors P, Wang Q, Ekelund M, Westrom BR, Jansson O, Lindstrom CL et al. Increased colonic permeability in patients with ulcerative colitis: an in vitro study. Scand J Gastroenterol 1998; 33(7):749-753.
(43) Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005; 129(2):550-564.
(44) Dubuquoy L, Jansson EA, Deeb S, Rakotobe S, Karoui M, Colombel JF et al. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology 2003; 124(5):1265-1276.
(45) Soderholm JD, Olaison G, Lindberg E, Hannestad U, Vindels A, Tysk C et al. Different intestinal permeability patterns in relatives and spouses of patients with Crohn's disease: an inherited defect in mucosal defence? Gut 1999; 44(1):96-100.
(46) May GR, Sutherland LR, Meddings JB. Is small intestinal permeability really increased in relatives of patients with Crohn's disease? Gastroenterology 1993; 104(6):1627-1632.
(47) Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A et al. Genetic basis for increased intestinal permeability in families with Crohn's disease: role of CARD15 3020insC mutation? Gut 2006; 55(3):342-347.
(48) Soderholm JD, Streutker C, Yang PC, Paterson C, Singh PK, McKay DM et al. Increased epithelial uptake of protein antigens in the ileum of Crohn's disease mediated by tumour necrosis factor alpha. Gut 2004; 53(12):1817-1824.
(49) Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol 2002; 97(8):2000-2004.
(50) Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M et al. Downregulation of epithelial apoptosis and barrier repair in active Crohn's disease by tumour necrosis factor alpha antibody treatment. Gut 2004; 53(9):1295-1302.
(51) Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE et al. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc Natl Acad Sci U S A 2005; 102(50):18129-18134.
(52) Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut 2004; 53(11):1658-1664.
(53) Wehkamp J, Wang G, Kubler I, Nuding S, Gregorieff A, Schnabel A et al. The Paneth cell alpha-defensin deficiency of ileal Crohn's disease is linked to Wnt/Tcf-4. J Immunol 2007; 179(5):3109-3118.
(54) Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF et al. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut 2003; 52(11):1591-1597.
(55) Keita A, Salim S, Jiang T, Yang PC, Franzen L, Soderkvist P et al. Increased uptake of non-pathogenic E. coli via the follicle-associated epithelium in longstanding ileal Crohn's disease. J Pathol 2008.
(56) Salim SY, Silva MA, Keita AV, Larsson M, Andersson P, Magnusson KE et al. CD83+CCR7- dendritic cells accumulate in the subepithelial dome and internalize translocated Escherichia coli HB101 in the Peyer's patches of ileal Crohn's disease. Am J Pathol 2009; 174(1):82-90.
(57) Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 2006; 25(2):309-318.
(58) Atreya I, Atreya R, Neurath MF. NF-kappaB in inflammatory bowel disease. J Intern Med 2008; 263(6):591-596.
(59) Zhou C, Tabb MM, Nelson EL, Grun F, Verma S, Sadatrafiei A et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest 2006; 116(8):2280-2289.
(60) Coste A, Dubuquoy L, Barnouin R, Annicotte JS, Magnier B, Notti M et al. LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc Natl Acad Sci U S A 2007; 104(32):13098-13103.