Sammanfattat
Fyra principiella patofysiologiska mekanismer avseende den perifera neuropatiska smärtan har betonats: 1. uttryck av aberranta jonkanaler, tex natriumkanaler, vilket skulle leda till spontan nervaktivitet, 2. uppreglering av kalciumkanaler med ökad mängd frisatt transmittor som resultat, 3. bombardemang av sk NMDA-receptorer postsynaptiskt i ryggmärgens bakhorn med hyperexcitabilitet som följd, 4. facilitering av impulsöverföringen i ryggmärgens bakhorn via nedåtstigande bansystem från hjärnstammen.
Vid beröringsorsakad smärta finns åtminstone i en del fall anledning att misstänka kommunikation mellan beröringssystemet och smärtsystemet, antingen i periferin (sk efaptisk transmission) eller i CNS.
Mekanismer bakom den centrala neuropatiska smärtans underhåll är till stora delar okända.
Kunskapsökningen inom området neuropatisk smärta har under de senaste två årtiondena varit påtaglig. Den prekliniska forskningen har sökt klarlägga mekanismer bakom den neuropatiska smärtans uppkomst och underhåll, medan den kliniska forskningen har koncentrerats kring karakteristik av symtom och statusfynd, och mer än hundra randomiserade, kontrollerade farmakologiska behandlingsstudier har publicerats.
Något besviket måste man konstatera att i ett behandlingsperspektiv har inga strategier mot neuropatisk smärta någonsin tagits fram med primärt mål att modifiera sådan smärta. De behandlingsalternativ som finns i dag, och som oftast endast ger partiell lindring, har hämtats från andra terapiområden. Orsakerna till detta tillkortakommande är nog flera, men de bottnar sannolikt i bristen på detaljerad kunskap om patofysiologiska mekanismer för den neuropatiska smärtans uppkomst och vidmakthållande.
Neuropatisk smärta – ny definition i vardande
IASP (International Association for the Study of Pain) definierar neuropatisk smärta som »pain initiated or caused by a primary lesion or dysfunction in the nervous system« [1]. Det har anmärkts att inklusionen av »dysfunction« i definitionen kan vara en källa till förvirring, eftersom det felaktigt kan tillåtas inkludera såväl nociceptiv, psykogen och etiologiskt okänd smärta som neuropatisk dito [2].
En arbetsgrupp inom IASPs intresseförening för neuropatisk smärta har nyligen publicerat en förnyad och mer preciserad definition av begreppet: »pain arising as a direct consequence of a lesion or disease affecting the somatosensory system« [3]. Man föreslog att definitionen skulle användas i forskning och klinisk praktik under några år för att därefter ånyo ifrågasättas. I Fakta återges ett antal neuropatiska tillstånd som är smärtsamma eller kan åtföljas av smärta.
Inblickar i några patofysiologiska mekanismer
Det smärtsamma lidandet vid neuropatisk smärta utmärks av såväl spontansmärta som stimulusinducerad smärta (Figur 1). De flesta patienter med neuropatisk smärta lider av spontan smärta, oftast ständigt pågående sådan med eller utan överlagrade paroxysmer eller aggravering med längre duration.
En minoritet har stimulusutlöst smärta, och här utgör allodyni (smärta vid normalt icke-smärtsam retning) det största problemet. Vanligen handlar det om smärta från lätt beröring av huden när retningen rör sig över huden alternativt vid lättare ihållande tryck. En del rapporterar köldallodyni, medan värmeallodyni sällan utgör ett problem.
Den klassiska trigeminusneuralgin utmärks av endast paroxysmal smärta med sekunders duration och helt smärtfria intervall dessemellan. Hos majoriteten av patienter med denna sjukdom kan man också utlösa smärtan vid beröring inom delar av det smärtande området, sk triggerzoner.
Djurmodeller har utvecklats för att försöka studera olika aspekter av de beskrivna kliniska symtomen och statusfynden. Vad som observeras i djurmodeller kan dock helt naturligt aldrig bli säkra återspeglingar av symtom och statusfynd hos patienter. Det pågår inom vetenskapssamhället en diskussion angående djurmodellernas relevans. Det finns ett stort antal djurmodeller som används för att studera neurofysiologiska, morfologiska och neurokemiska förändringar, som antas bidra till utvecklingen av neuropatirelaterade symtom och känselförändringar. Särskilt omfattande användning kan noteras för modeller med olika typer av traumatisk ischiadicusskada [4].
Det är dock uppenbart att många av dessa neuropatimodeller har en etiologi som påtagligt avviker från det man ser i kliniken. Djurmodeller används för att studera, såväl perifert som centralt, mekanismer som kan tex förstärka eller förlänga resultatet av en given retning. Djurexperimentella studier kring möjligheten av att spontansmärta föreligger förekommer endast undantagsvis. Majoriteten av studierna är inriktade på beteendeförändringar vid givna retningar före och efter nervskada, men även tex elektrofysiologisk registrering av nervcellsaktivitet i ryggmärgens bakhorn används för att studera spontanaktivitet (möjligen relaterat till upplevelse av spontansmärta) och stimulusutlöst aktivitet. Molekylära, immunhistokemiska och, på senare tid, genetiska tekniker har också en utbredd användning inom området.
Att försöka korrelera sannolikt smärtrelaterade beteendeparametrar i djurmodellerna med förändringar i jonkanaler, receptorer etc är en viktig framtida uppgift för forskarsamhället. Försöksdjurens genetiska bakgrund förefaller också spela roll för utvecklingen av tecken på möjlig neuropatisk smärta och svar på behandling [5, 6].
Endast en fraktion av individer med nervskada utvecklar smärtproblem, och av dessa kan vi med till buds stående behandlingsstrategier sannolikt hjälpa cirka hälften med betydelsefull smärtlindring. Djurexperimentella studier antyder betydligt bättre behandlingsutfall. Kliniskt finns en rad variabler som kan försvåra behandlingen, alltifrån följsamhet avseende behandlingen, tillståndets duration till affektiva komponenter i upplevelsen. De senare är uttalat svåra att värdera i djurmodeller. Modellerna ger inte heller information kring biverkningar såsom yrsel, mardrömmar, kognitiva störningar eller hallucinationer, vilka kan vara avgörande kliniska problem.
Perifer neuropatisk smärta
Inga anspråk görs här på att belysa samtliga möjliga patofysiologiska mekanismer, eftersom de flesta av dem i dagsläget inte kan översättas till symtom och/eller statusfynd hos patienter. I stället fokuseras på sådana mekanismer som förefaller återkomma i upprepade djurexperimentella neuropatimodeller.
Mekanismer bakom spontan pågående och intermittent/ paroxysmal smärta samt stimulusinducerad smärta inbegriper sannolikt ökad aktivitet i primära afferenter. Denna aktivitetsökning orsakas rimligen av ökad spontan impulsaktivitet inkluderande sk ektopisk impulsbildning (signaler som uppstår vid skadeplatsen) och efaptisk transmission (direkt elektrisk koppling mellan perifera neuron till följd av dålig elektrisk isolering efter nervskada, »överhörning«). Sådana mekanismer är sannolikt betydelsefulla för smärtgenerering vid nervskador perifert om dorsalrotsgangliet.
Vid axotomiskador på nervrötter kan sannolikt ökad aktivitet i det centrala nervsystemet föreligga som ett uttryck för disinhibition (bortfall av inhibition medför excitation) och/eller spontant aktiva hyperexcitabla spinala/trigeminala neuron. Den enskilda patofysiologiska faktorns betydelse för uppkomst av smärta känner man i dag inte till. Möjligheten att vissa kombinationer av förändringar bildar den nödvändiga konstellationen för smärtans uppkomst och underhåll måste också beaktas.
Det har under senare tid framförts att perifer nervskada skulle kunna liknas vid en neurodegenerativ sjukdom med en central roll för degenerativa förändringar i ryggmärgens bakhorn för smärtuppkomst och underhåll [Clifford Woolf, Boston, muntlig presentation vid IASPs världskongress 2005, Sydney]. Mot betydelsen av detta, åtminstone för spontansmärtan, talar det faktum att majoriteten av patienter med nervskador perifert om dorsalrotsgangliet blir smärtfria vid en ledningsblockad med lokalanestetika given proximalt om skadenivån [7].
Smärtmekanismer visade i djurmodeller för perifer neuropati
Hundratals neurofysiologiska, morfologiska och neurokemiska förändringar har påvisats i olika djurmodeller för neuropati och förmodad neuropatisk smärta under de senaste cirka 20 åren. Möjligheten av att fynden är relaterade till nervskadan och inte den eventuellt förekommande smärtan måste beaktas. Man har nyligen via resultat från djurmodeller för neuropati försökt identifiera perifera och centrala mekanismer, vilka antas vara av särskild betydelse för den kliniska perifera neuropatiska smärtans uppkomst och underhåll, och dessa förefaller vara gemensamma för många av de nu använda modellerna för perifer neuropati och förmodad neuropatisk smärta [8, 9]. Dessa förändringar kan återfinnas isolerade eller i olika kombinationer (Figur 2):
Förändringar i generering av aktionspotentialer (dvs nervsignaler) genom att nervcellens uttryck av jonkanaler, framför allt natriumkanaler, ger underlag för spontan och faciliterad stimulusinducerad nervaktivitet vid och i närheten av nervskadan samt i området för dorsalrotsgangliet [10, 11].
Förändringar i transmittorfrisättning från nociceptiva afferenter genom ökat uttryck av och förändrad funktion hos presynaptiska kalciumkanaler [12-14].
Excitabilitetsökning hos neuron i ryggmärgens bakhorn/ spinala trigeminuskärnan [15].
Ökad descenderande facilitering från hjärnstammen eller förändrad balans mellan nedåtstigande facilitering och inhibition [16-18].
Förändringar i jonkanaler. Efter ischiadicusskada uppkommer spontanaktivitet i perifera afferenter [19]. Med mikroneurografisk registreringsteknik har man hos människa observerat förekomst av sådan spontanaktivitet som skulle kunna utgöra underlag för upplevelser av sensoriska symtom [20]. Man antar också att ektopisk aktivitet kan bidra till utveckling av känslighetsökning (sensitisering) i CNS och därmed ett hyperexcitabelt tillstånd som kan ligga bakom tex olika former av allodyni. Mer specifikt har det föreslagits att det neuronala bombardemanget från särskilt omyeliniserade C-fibrer är orsaken till sådan hyperexcitabilitet [21-23]. Normalt förekommer ingen kommunikation mellan olika sensoriska nervfibrer i den perifera nerven. Efter axotomi kan såväl elektriskt som kemiskt medierad aktivering av närliggande neuron bli möjlig, sk efaptisk transmission [24]. Genom sådana mekanismer kan det patologiska inflödet i perifera nerver efter nervskada involvera fler nervtrådar än bara de som är primärt skadade.
Stort intresse har utvecklats kring konceptet att natriumkanaler skulle vara en lämplig måltavla för framtida läkemedel mot neuropatisk smärta, eftersom det nu klarlagts att uttrycket av sådana jonkanaler ökar efter nervskada [25]. Det finns i dag 10 kända typer av natriumkanaler, där framför allt de sk Nav1.3, Nav1.7 och Nav1.8 (spänningsberoende natriumkanaler) tilldragit sig intresse i detta sammanhang. Glial-derived growth factor (GDNF), som visats normalisera förmodat beteende relaterat till neuropatisk smärta hos försöksdjur, normaliserar också uttycket av Nav1.3 [26]. Data har också publicerats som antyder betydelse av Nav1.8 [27].
Förändringar i kalciumkanaler. Synapsnära förstärkning av resultatet av inkommande signaler i den primära afferenten har hypotetiskt framförts som en möjlig strategi för nervsystemet att kompensera för ett bortfall av inflöde i samband med en partiell perifer nervskada [15]. Presynaptiskt skulle detta kunna ske genom ökad frisättning av transmittorsubstans i samband med att aktionspotentialen invaderar den primära afferentens terminal i ryggmärgens bakhorn.
Spänningsaktiverade kalciumkanaler (av sk N-typ), aktiverade av kraftig membrandepolarisation, är betydelsefulla i smärtbanorna. Särskilt kan här nämnas deras betydelse för transmittorfrisättning från den primära afferentens terminal.
Jonkanalerna är alla sammansatta av en sk (alfa)1-underenhet, som bildar kanalens por, men även av (beta)-, (alfa)2(delta)- och c-subenheter (proteiner), och man har påvisat en uppreglering av sådana kanaler efter nervskada [13]. Betydelsen av dessa kalciumkanaler får också stöd av neuropatistudier utförda på mutanta möss som saknar N-typen av kalciumkanaler. Sådana djur visade mindre tecken på överkänslighet än en kontrollgrupp [28].
Andra studier har nyligen pekat på en uppreglering av (alfa)2(delta)-subenheten och ökad känslighet för läkemedlen gabapentin och pregabalin efter diabetesinducerad respektive mekaniskt inducerad neuropati. Uppregleringen har föreslagits vara orsaken till överkänsligheten för perifer retning i dessa modeller [12].
Funktionsförändringar i ryggmärgsneuron. Nervskada är normalt associerad med förlust av sensoriska nervfibrer [29]. I ett sådant scenario är det rimligt att anta att närvaron av stimulusinducerad överkänslighet är ett resultat av centrala förändringar som medför att reducerat inflöde från skadade nerver kompenseras [29]. Perifera mekanismer såsom efaptisk transmission/»överhörning« kan inte uteslutas ha viss betydelse i detta sammanhang. Ökade svar från spinala neuron på vissa retningar kan vara en konsekvens av ökad central excitabilitet, tex central sensitisering som i sin tur inducerats av nervskadeinducerad ektopisk aktivitet på grund av förändring i jonkanalsuttryck [29]. I det sammanhanget kan man notera att avledning från ryggmärgsneuron har indikerat påtaglig central plasticitet efter nervskada med förstorade receptoriska fält och förändrade urladdningsmönster [29, 30].
En av receptorerna som återfinns på bakhornsneuron i ryggmärgen, den sk NMDA-receptorn, har egenskaper som gör den till en nyckelstruktur avseende centrala mekanismer vid hyperexcitabilitet [31]. En av de mer intressanta egenskaperna är dess medverkan i uppkomsten av »wind-up« efter kraftig repetitiv nociceptiv retning [32].
»Wind-up« beskriver den successiva ökningen i nervsvar vid en repetitiv retning av C-fibrer (Figur 3) [33, 34], som resulterar i frisättning av excitatoriska aminosyror och peptider i dorsalhornets ytliga lager, vilka i sin tur orsakar depolarisation av det postsynaptiska neuronet. Rekryteringen av NMDA-receptorer förstärks av kväveoxidproduktion och så småningom via geninduktion i ryggmärgsneuronen [33, 35].
Man känner till att excitatoriska aminosyror som verkar på NMDA-receptorn bidrar till utvecklingen av den ökade retbarheten i ryggmärgsneuron [36]. Uppreglering av receptorer för excitatoriska aminosyror i ipsilaterala dorsalhornet har påvisats efter perifer nervskada, vilket pekar mot att NMDA-receptorer bidrar till utveckling av förändrad funktion [37]. Elektrofysiologiska studier som visar att »wind-up« signifikant reduceras efter applikation av NMDA-receptorantagonister konfirmerar NMDA-receptorns roll i fenomenet [38].
Vid perifer nervskada kan transganglionär axonal degeneration in i ryggmärgen, alternativt in i spinala trigeminuskärnan i hjärnstammen, sannolikt ge upphov till omställningar av den synaptiska harmonin med konsekvenser för balansen mellan excitatoriskt och inhibitoriskt inflöde, vilket rimligen kan ha betydande följdverkningar. Om bortfallet framför allt drabbar grova myeliniserade fibrer, kommer möjligheten till modulering av inflödet i nociceptiva afferenter via mekanoreceptiva trådar att störas, sk disinhibition [39, 40].
Vidare kan denna reduktion av inhibitorisk kontroll möjligen också leda till spontanaktivitet i nociceptiva spinala neuron. Spontanaktivitet i sådana neuron kan sannolikt också uppkomma som ett resultat av excitatoriska aminosyrors toxiska inverkan på små inhibitoriska interneuron i bakhornet, som därmed kan gå under [41].
Vidare kan deafferentering genom rotavskärning (jämför med traumatisk avulsion av tex delar av brakialplexus) leda till hyperaktivitet i bakhornsneuron [42] och på så sätt generera spontansmärta.
Ett uttryck för att perifer nervskada kan leda till reducerad inhibitorisk aktivitet i ryggmärgens bakhorn är fyndet att GABA(gammaaminosmörsyra)-förekomsten är reducerad i ryggmärgens ytliga delar efter nervskada [43] och att GABA-B-receptoragonistbindning i ryggmärgens lamina II minskar 2–4 veckor efter nervlesion [44]. Det sistnämnda kan möjligen förklara den kliniska observationen att åtminstone peroral baklofenbehandling, en GABA-B-agonist, oftast är ineffektiv vid kronisk neuropatisk smärta. Andra farmakologiska studier har påvisat ökad GABAerg inhibitorisk aktivitet av spinala neuron efter perifer nervskada. Man föreslår att detta skulle kunna vara ett uttryck för kompensatoriska mekanismer för att balansera den ökade excitabiliteten efter nervskada [45].
Ökad descenderande facilitering. Nyligen har föreslagits att descenderande facilitering utgående från hjärnstammen skulle vara ett bidragande fenomen för underhåll av neuropatisk smärta. NK-1-receptorer (neurokinin) hos bakhornsneuron som exciteras av substans P skulle utgöra början på en spino-bulbo-spinal loop som har betydelse för excitabilitet i dorsalhornsneuron. Majoriteten av NK-1-uttryckande neuron i lamina I av ryggmärgen ascenderar till hjärnstamsregioner såsom det parabrakiala området och den periakveduktala grå substansen samt till talamus; till den sistnämnda särskilt till områden som projicerar på storhjärnsområden associerade med affektiva och kognitiva funktioner [46, 47]. Förändringar i dessa neuronkretsar har föreslagits ligga bakom plastiska förändringar som inducerats av neuropati.
Försök med substans P-saporin (SP-SAP), som binder selektivt till NK-1-uttryckande neuron i lamina I/III, har visat en markerad minskning i retningsöverkänslighet vid beteendestudier efter nervskada, vilket sker parallellt med reducerad excitabilitet i bakhornsneuron [48, 49]. Möjligheten att reproducera många av de elektrofysiologiska förändringarna genom farmakologisk blockering av spinala 5HT3-receptorer frambringade hypotesen att NK-1-uttryckande lamina I/III-neuron befinner sig i början av en spino-bulbo-spinal loop, som driver serotonerg excitatorisk influens från hjärnstammen [49].
Detta kan bidra till förståelsen av andra nyligen presenterade resultat talande för betydelsen av descenderande facilitering för uttryck av smärttillstånd, inkluderande de neuropatiska [16, 17, 50]. En ökad funktionell roll för denna excitatoriska descenderande bana har demonstererats efter nervskada [18], och administration av 5HT3-receptorantagonisten ondansetron reverserar överkänslighet för mekanisk retning efter ryggmärgsskada på råtta [51]. Ondansetron har också rapporterats ge smärtlindring vid kliniska försök på patienter med förmodad neuropatisk smärta [52].
Sammantaget kan NK-1-uttryckande neuron i lamina I, som påverkats av förändrad aktivitet i skadade primära afferenter orsakad av natrium- och kalciumkanalsstörningar samt via den ovan beskrivna spino-bulbo-spinala loopen, bidra till förändrad transmission i ryggmärgens bakhorn.
Allodyni
Endast de vanligaste formerna av mekanisk allodyni ska beskrivas här, sk beröringsallodyni (dynamic mechanical allodynia) och tryckallodyni (static mechanical allodynia).
Dynamisk mekanisk allodyni, den sannolikt kliniskt mest frekvent förekommande typen av mekanisk allodyni, framkallas genom en mjuk strykning över huden och statisk mekanisk allodyni genom ihållande lätt tryck. Baserat på experimentella kompressions-/ischemistudier har det föreslagits att den dynamiska mekaniska allodynin medieras av myeliniserade afferenter [53], medan statisk mekanisk allodyni medieras av omyeliniserade afferenter [54]. Fortsättningsvis diskuteras endast den kunskap som finns om den dynamiska mekaniska allodynin.
Dynamisk mekanisk allodyni kan vara extremt lokaliserad hos patienter med nervskada och neurombildning av nociceptiva fibrer, men den är oftare spridd inom hela eller delar av den skadade nervens kutana innervationsområde. Det finns en rad fynd från humanlitteraturen som ger stöd åt betydelsen av A-betafibrer som den initiala budbäraren vid dynamisk mekanisk allodyni:
• Man har visat att latensen mellan mekanisk retning och rapporterad sensation indikerar involvering av snabbt ledande A-betafibrer [55, 56].
• Från studier med kompression/ischemiinducerad blockad av nerver har rapporterats att dynamisk mekanisk allodyni lindras när funktionen i A-beta-/A-deltafibrer upphört [53, 57].
• Lokalanestetika som endast blockerar A-delta- och C-fibrer påverkar inte dynamisk mekanisk allodyni hos patienter med neuropatisk smärta [57, 58].
• Första sensationen av TENS var smärtsam hos patienter med beröringsallodyni, vilket indikerar att aktivering av A-betafibrer, som till följd av sin tjocklek har lågt longitudinellt motstånd, kan vara smärtsam [59].
Med iakttagande av en kritisk hållning till djurmodellernas kliniska relevans ger den djurexperimentella litteraturen stöd åt en rad möjliga patofysiologiska mekanismer för uppkomst och underhåll av dynamisk mekanisk allodyni:
• Perifer sensitisering av A-delta-/C-fibrer. Hur vanligt detta är vid långvarig neuropatisk smärta är inte känt men det förekommer hos undergrupper av patienter med postherpetisk neuralgi [60]. Förekomsten vid andra perifera neuropatiska smärtor är okänd.
• Aktivering av »tysta« nociceptorer [61].
• Efaptisk transmission. I en skadad nerv kan isoleringen mellan olika ingående nervtrådar gå förlorad, vilket möjliggör direkt elektrisk aktivering av närliggande fibrer. Sådan sk efaptisk transmission mellan A-betafibrer och nociceptiva afferenter har rapporterats, men är sannolikt sällsynt förekommande [62].
• Förlust av A-betamedierad inhibition i dorsalhornet [63] med störd balans mellan excitation och inhibition.
• Central sensitisering, dvs öppnande av tidigare existerande men funktionellt tysta synaptiska kontakter mellan grova myeliniserade afferenter och nociceptivt specifika neuron i ryggmärgens bakhorn [64]. Detta är sannolikt en fysiologisk mekanism som uttrycks i området för sekundär hyperalgesi (allodyni) vid kirurgiska eller andra skador och i experimentella smärtmodeller [65, 66]. Om denna mekanism inte normaliseras finns underlag för ihållande beröringsallodyni.
• Tonisk aktivitet i descenderande faciliterande system från hjärnstammen (se ovan).
• »Sprouting« av mekanoreceptiva fibrer i ryggmärgens bakhorn vilka etablerar nya synaptiska kontakter med nociceptiva neuron har diskuterats, men förefaller mindre betydelsefullt än vad man initialt antog [67, 68].
Dysestesi
Föga uppmärksamhet har ägnats dysestesi, dvs obehag i samband med retning inom det neuropatiska området, ett inte ovanligt fenomen vid neuropatisk smärta. Sannolikt beror detta på att djurexperimentella studier inte är applicerbara, eftersom perceptuella detaljer inte kan kontrolleras. Studier med sk mikroneurografisk teknik på människa har föreslagit att multiplikation av impulser i skadade grova myeliniserade A-betafibrer kan ge en upplevelse av obehag [69].
Storhjärnans aktivitet vid perifer neuropatisk smärta
Undersökningar av hjärnbarkens aktivitet hos människa med positronemissionstomografiteknik (PET) har använts för att visualisera aktivitet i specifika hjärnområden vid klinisk smärta, och ett flertal studier har publicerats från Karolinska institutet. Vid kronisk smärtsam mononeuropati rapporterades intressant nog ingen ökad aktivitet i primära (S1) och sekundära (S2) somatosensoriska kortex [70]. Särskilt iögonfallande var i stället fyndet av ökad aktivitet i högra sidans anteriora gyrus cinguli (Brodmanns area [BA] 24) samt bilateralt (oavsett om smärtan återfanns på höger eller vänster sida) i insula (BA14), prefrontalkortex (BA10 och 47) och parietalkortex (BA 7 och 40). Resultaten från patienter med kronisk neuropatisk smärta påvisar således aktivitet i framför allt hjärnbarksområden som knyts till affektiva komponenter av smärtupplevelsen.
Det centralnervösa processandet av upplevelsen kopplad till dynamisk mekanisk allodyni hos patienter med smärtsam perifer traumatisk neuropati har också studerats med PET-teknik [71]. Till skillnad från vad som beskrivits om spontansmärta kunde man här påvisa en omfattande aktivering av S1 och S2. Aktiveringen av S1 kontralateralt till stimuleringsområdet var påtagligt mer omfattande till såväl styrka som utbredning än endast beröringsupplevelse vid retning kontralateralt till allodynin. Jämfört med fynden vid aktivering av det sk mediala smärtsystemet vid spontan pågående perifer neuropatisk smärta såg man således en påtaglig aktivering av det laterala smärtsystemet vid dynamisk mekanisk allodyni.
Central neuropatisk smärta
Med central neuropatisk smärta avses smärta orsakad av lesion eller sjukdom i centrala nervsystemet. Sådan smärta kan uppstå efter skada/vid sjukdom utefter hela CNS, dvs från ryggmärgens bakhorn till hjärnbarken. Kunskapsfältet kring central smärta är i alla delar mycket begränsat vid en jämförelse med kunskapen om perifer neuropatisk smärta. Endast ett fåtal djurexperimentella modeller har presenterats, de flesta med olika typer av ryggmärgsskada. Betydelsen av dessa för ökad förståelse av de två huvudtyperna av central neuropatisk smärta efter ryggmärgsskada, övergångszonssmärta respektive smärta nedanför skadenivån, är ännu oklar.
Några av de mest omhuldade teorierna bakom uppkomsten av central neuropatisk smärta är följande:
• Irritabelt fokus vid skadestället [72].
• Lesion av mediala lemnisken ledande till disinhibition av smärtsystemet [73].
• Lesion i den spino(trigemino)-talamo-kortikala banan [74, 75].
• Förlust av köldaktiverade spino-talamiska projektionsneuron ledande till disinhibition [76]
• Förlust av inhibitorisk influens från retikulära talamuskärnan [77, 78].
Såväl supra- som infratentoriala lesioner kan orsaka denna typ av smärta, vilket tydligt talar mot en fortsatt användning av begreppet »talamussyndrom/talamussmärta« som introducerades av Dejerine och Roussy i början av förra seklet. Talamus per se behöver inte vara involverad för att centrala neuropatiska smärttillstånd ska uppkomma efter infarkt/blödning i CNS [75]. Av anatomiska skäl är dock till talamus inkommande eller från talamus avgående nervfibrer ofta involverade vid infarkt/blödning, vilket troligen är skälet till att begreppet »talamussmärta« fortlever. Central neuropatisk smärta synes kunna uppstå vid en rad olika tillstånd, såsom vaskulära lesioner, traumatiska ryggmärgsskador, multipel skleros och syringomyeli.
Det står klart att störning av det spino(trigemino)-talamo-kortikala bansystemet är en förutsättning för att utveckla central neuropatisk smärta [75, 79, 80]. Det är viktigt att notera att inte alla patienter med sådan störning utvecklar central neuropatisk smärta. Vilka ytterligare faktorer som krävs för smärtutveckling känner vi i dag inte till. Kanske är skadeomfattningen betydelsefull eller kanske är samtidiga störningar av andra bansystem en nödvändig förutsättning.
*
Potentiella bindningar eller jävsförhållanden: Författaren har under det senaste året fungerat som rådgivare åt Pfizer, Lilly, Neurosearch, Boehringer Ingelheim, Nolabs, Wyeth och Medtronic samt erhållit föreläsningsarvoden från Pfizer, Lilly och Boehringer Ingelheim.
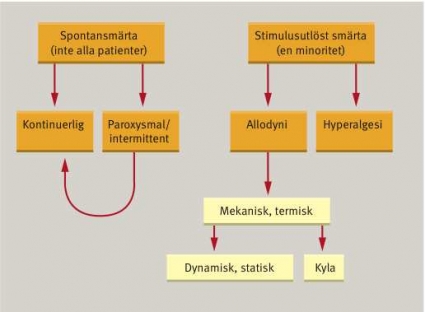
Figur 1. Symtom och statusfynd vid neuropatisk smärta.
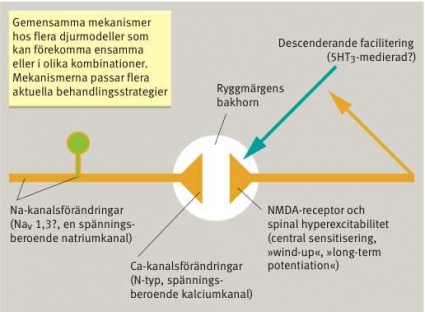
Figur 2. Föreslagna vanliga mekanismer vid perifer neuropatisk smärta.

Figur 3. Schematisk återgivning av »wind-up«-fenomenet. I figuren återges registrering av aktivitet från ryggmärgsneuron vid perifer elektrisk retning som aktiverar C-fibrer. Notera den tilltagande depolariseringen av membranpotentialen samt den ökande impulsfrekvensen och ökningen av impulsskurens duration vid upprepade retningar.
Referenser
1. Merskey H, Bogduk N, eds. Classification of chronic pain: Descriptions of chronic pain syndromes and definitions of pain terms. 2nd ed. Seattle: IASP Press; 1994, p. 222.
2. Hansson P, Lacerenza M, Marchettini P. Aspects of clinical and experimental neuropathic pain: The clinical perspective. In: Hansson PT, Fields HL, Hill RG, Marchettini P. Neuropathic pain: Pathophysiology and treatment. Seattle: IASP Press; 2001. p. 1-18.
3. Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70(18):1630-5.
4. Kim KJ, Yoon YW, Chung JM. Comparison of three rodent neuropathic pain models. Exp Brain Res. 1997;113(2):200-6.
5. Mogil JS, Nessim LA, Wilson SG. Strain-dependent effects of supraspinal orphanin FQ/nociceptin on thermal nociceptive sensitivity in mice. Neurosci Lett. 1999;261(3):147-50.
6. Chesler EJ, Ritchie J, Kokayeff A, Lariviere WR, Wilson SG, Mogil JS. Genotype-dependence of gabapentin and pregabalin sensitivity: the pharmacogenetic mediation of analgesia is specific to the type of pain being inhibited. Pain. 2003;106(3):325-35.
7. Arnér S, Lindblom U, Meyerson BA, Molander C. Prolonged relief of neuralgia after regional anesthetic blocks. A call for further experimental and systematic clinical studies. Pain. 1990;43(3):287-97.
8. Hansson PT, Dickenson AH. Pharmacological treatment of peripheral neuropathic pain conditions based on shared commonalities despite multiple etiologies. Pain. 2005;113(3):251-4.
9. Dickenson AH, Matthews EA, Suzuki R. Neurobiology of neuropathic pain: mode of action of anticonvulsants. Eur J Pain. 2002;6 Suppl A:51-60.
10. Devor M. The pathophysiology of damaged peripheral nerves. In: Wall PD MR. Textbook of pain. Edinburgh: Churchill Livingstone; 1994. p. 79-100.
11. Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci U S A. 1999;96(14):7635-9.
12. Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, et al. . Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther. 2002;303(3):1199-205.
13. Matthews EA, Dickenson AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92(1-2):235-46.
14. Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, Orendacova J, et al. . Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp Brain Res. 2002;147(4):456-63.
15. Dickenson A, Matthews E, Suzuki R. Central nervous system mechanisms of pain in peripheral neuropathy. In: Hansson PT, Fields HL, Hill RG, Marchettini P. Neuropathic pain: pathophysiology and treatment. Seattle: IASP Press; 2001. p. 85-106
16. Pertovaara A. A neuronal correlate of secondary hyperalgesia in the rat spinal dorsal horn is submodality selective and facilitated by supraspinal influence. Exp Neurol. 1998;149(1):193-202.
17. Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25(6):319-25.
18. Suzuki R, Rahman W, Hunt SP, Dickenson AH. Descending facilitatory control of mechanically evoked responses is enhanced in deep dorsal horn neurones following peripheral nerve injury. Brain Res. 2004;1019(1-2):68-76.
19. Devor M, Raber P. Heritability of symptoms in an experimental animal model of neuropathic pain. Pain. 1990;42:51-67.
20. Nordin M, Nyström B, Wallin U, Hagbarth K-E. Ectopic sensory discharges and paraethesiae in patients with disorders of peripheral nerves, dorsal roots and dorsal columns. Pain. 1984;20:231-245.
21. Wu G, Ringkamp M, Hartke TV, Murinson BB, Campbell JN, Griffin JW, et al. . Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci. 2001;21(8):RC140.
22. Woolf CJ, Decosterd I. Implications of recent advances in the understanding of pain pathophysiology for the assessment of pain in patients. Pain. 1999;Suppl 6:S141-7.
23. Ma QP, Woolf CJ. Progressive tactile hypersensitivity: An inflammation-induced incremental increase in the excitability of the spinal cord. Pain. 1996;67:97-106.
24. Devor M, Wall PD. Cross excitation among dorsal root ganglion neurons in nerve injured and intact rats. J Neurophysiol. 1990;64:1733-1746.
25. Waxman SG, Cummins TR, Dib-Hajj S, Fjell J, Black JA. Sodium channels, excitability of primary sensory neurons, and the molecular basis of pain. Muscle Nerve. 1999;22(9):1177-87.
26. Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB. Potent analgesic effects of GDNF in neuropathic pain states. Science. 2000;290(5489):124-7.
27. Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, et al. . Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95(1-2):143-52.
28. Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T, et al. . Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. Embo J. 2001;20(10):2349-56.
29. Suzuki R, Dickenson AH. Neuropathic pain: nerves bursting with excitement. Neuroreport. 2000;11(12):R17-21.
30. Suzuki R, Kontinen VK, Matthews E, Williams E, Dickenson AH. Enlargement of the receptive field size to low intensity mechanical stimulation in the rat spinal nerve ligation model of neuropathy. Exp Neurol. 2000;163(2):408-13.
31. Dickenson AH, Sullivan AF. Differential effects of excitatory amino acid antagonists on dorsal horn nociceptive neurones in the rat. Brain Res. 1990;(506):31-39.
32. Dickenson AH. A cure for wind up: NMDA receptor antagonists as potential analgesics. Trends Pharmacol Sci. 1990;11(8):307-9.
33. Dickenson AH. Central acute pain mechanisms. Ann Med. 1995;27(2):223-7.
34. Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain. 1999;79(1):75-82.
35. Stanfa LC, Misra C, Dickenson AH. Amplification of spinal nociceptive transmission depends on the generation of nitric oxide in normal and carrageenan rats. Brain Res. 1996;737(1-2):92-8.
36. Coderre TJ, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12(9):3665-70.
37. Harris JA, Corsi M, Quartaroli M, Arban R, Bentivoglio M. Upregulation of spinal glutamate receptors in chronic pain. Neuroscience. 1996;74(1):7-12.
38. Suzuki R, Matthews EA, Dickenson AH. Comparison of the effects of MK-801, ketamine and memantine on responses of spinal dorsal horn neurones in a rat model of mononeuropathy. Pain. 2001;91(1-2):101-9.
39. Melzack R, Wall PD. Pain mechanisms: A new theory. Science. 1965;(150):971-978.
40. Woolf CJ, Wall PD. Chronic peripheral nerve section diminishes the primary afferent A-fibre mediated inhibition of rat dorsal horn neurones. Brain Res. 1982;242(1):77-85.
41. Sugimoto T, Bennett GJ, Kajander KC. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Peptides. 1990;11(4):205-13.
42. Lombard M-C, Nashold BS, Albe-Fessard D, Salman N, Sukr C. Deafferentation hypersensitivity in the rat after dorsal rhizotomy: a possible animal model of chronic pain. Pain. 1979;6:163-174.
43. Castro-Lopes JM, Tavares I, Coimbra A. GABA decreases in the spinal cord dorsal horn after peripheral neurectomy. Brain Res. 1993;620(2):287-91.
44. Castro-Lopes JM, Malcangio M, Pan BH, Bowery NG. Complex changes of GABAA and GABAB receptor binding in the spinal cord dorsal horn following peripheral inflammation or neurectomy. Brain Res. 1995;679(2):289-97.
45. Kontinen VK, Stanfa LC, Basu A, Dickenson AH. Electrophysiologic evidence for increased endogenous gabaergic but not glycinergic inhibitory tone in the rat spinal nerve ligation model of neuropathy. Anesthesiology. 2001;94(2):333-9.
46. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
47. Todd AJ, McGill MM, Shehab SA. Neurokinin 1 receptor expression by neurons in laminae I, III and IV of the rat spinal dorsal horn that project to the brainstem. Eur J Neurosci. 2000;12(2):689-700.
48. Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, et al. . Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278(5336):275-9.
49. Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002;5(12):1319-26.
50. Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66(6):355-474.
51. Oatway MA, Chen Y, Weaver LC. The 5-HT3 receptor facilitates at-level mechanical allodynia following spinal cord injury. Pain. 2004;110(1-2):259-68.
52. McCleane GJ, Suzuki R, Dickenson AH. Does a single intravenous injection of the 5HT3 receptor antagonist ondansetron have an analgesic effect in neuropathic pain? A double-blinded, placebo-controlled cross-over study. Anesth Analg. 2003;97(5):1474-8.
53. Ochoa JL, Yarnitsky D. Mechanical hyperalgesias in neuropathic pain patients: dynamic and static subtypes. Ann Neurol. 1993;33(5):465-72.
54. Cline MA, Ochoa J, Torebjork HE. Chronic hyperalgesia and skin warming caused by sensitized C nociceptors. Brain. 1989:621-47.
55. Fruhstorfer H, Lindblom U. Sensibility abnormalities in neuralgic patients studied by thermal and tactile pulse stimulation. In: von Euler C, Franzén O, Lindblom U, Ottoson D. Somatosensory mechanisms. London: Pitman Press; 1984. p. 353-361
56. Lindblom U, Verrillo RT. Sensory functions in chronic neuralgia. J Neurol Neurosurg Psychiatry. 1979;42(5):422-35.
57. Campbell JN, Raja SN, Meyer RA, Mackinnon SE. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32(1):89-94.
58. Nurmikko T, Wells C, Bowsher D. Pain and allodynia in postherpetic neuralgia: role of somatic and sympathetic nervous systems. Acta Neurol Scand. 1991;84(2):146-52.
59. Price DD, Bennett GJ, Rafii A. Psychophysical observations on patients with neuropathic pain relieved by a sympathetic block. Pain. 1989;36(3):273-288.
60. Fields HL, Rowbotham M, Baron R. Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiol Dis. 1998;5(4):209-27.
61. Schmidt R, Schmelz M, Forster C, Ringkamp M, Torebjork E, Handwerker H. Novel classes of responsive and unresponsive C nociceptors in human skin. J Neurosci. 1995;15(1 Pt 1):333-41.
62. Amir R, Devor M. Axonal cross-excitation in nerve-end neuromas: comparison of A- and C-fibers. J Neurophysiol. 1992;68(4):1160-6.
63. Laird JM, Bennett GJ. Dorsal root potentials and afferent input to the spinal cord in rats with an experimental peripheral neuropathy. Brain Res. 1992;584(1-2):181-90.
64. Cook AJ, Woolf CJ, Wall PD, McMahon SB. Dynamic receptive field plasticity in rat spinal cord dorsal horn following C-primary afferent input. Nature. 1987;325(7000):151-3.
65. Koltzenburg M, Lundberg LE, Torebjörk HE. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1992;51(2):207-219.
66. Torebjörk HE, Lundberg LE, LaMotte RH. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol (Lond). 1992;448(765):765-780.
67. Woolf CJ, Shortland P, Coggeshall RE. Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature. 1992;355(6355):75-8.
68. Bao L, Wang HF, Cai HJ, Tong YG, Jin SX, Lu YJ, et al. Peripheral axotomy induces only very limited sprouting of coarse myelinated afferents into inner lamina II of rat spinal cord. Eur J Neurosci. 2002;16(2):175-85.
69. Serra J, Ochoa J, Campero M. Human studies of primary nociceptors in neuropathic pain. In: Hansson PT, Fields HL, Hill RG, Marchettini P. Neuropathic pain: Pathophysiology and treatment. Seattle: IASP Press.; 2001. p. 63-83
70. Hsieh JC, Belfrage M, Stone-Elander S, Hansson P, Ingvar M. Central representation of chronic ongoing neuropathic pain studied by positron emission tomography. Pain. 1995;63(2):225-36.
71. Petrovic P, Ingvar M, Stone-Elander S, Petersson KM, Hansson P. A PET activation study of dynamic mechanical allodynia in patients with mononeuropathy. Pain. 1999;83(3):459-70.
72. Dejerine J, Roussy G. La syndrome thalamique. Rev Neurol. 1906;(14):521-532.
73. Head H, Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;(34):102-254.
74. Beric A, Dimitrijevic MR, Lindblom U. Central dysesthesia syndrome in spinal cord injury patients. Pain. 1988;34(2):109-16.
75. Boivie J, Leijon G, Johansson I. Central post-stroke pain–a study of the mechanisms through analyses of the sensory abnormalities. Pain. 1989;37(2):173-85.
76. Craig AD. A new version of the thalamic disinhibition hypothesis of central pain. Pain forum. 1998;(7):1-14.
77. Schott B, Laurent B, Mauguiere F. [Thalamic pain: critical study of 43 cases]. Rev Neurol (Paris). 1986;142(4):308-15.
78. Mauguiere F, Desmedt JE. Thalamic pain syndrome of Dejerine-Roussy. Differentiation of four subtypes assisted by somatosensory evoked potentials data. Arch Neurol. 1988;45(12):1312-20.
79. Vestergaard K, Nielsen J, Andersen G, Ingeman-Nielsen M, Arendt-Nielsen L, Jensen TS. Sensory abnormalities in consecutive, unselected patients with central post-stroke pain. Pain. 1995;61(2):177-86.
80. Svendsen KB, Jensen TS, Hansen HJ, Bach FW. Sensory function and quality of life in patients with multiple sclerosis and pain. Pain. 2005;114(3):473-81.