Sammanfattat
Nya cancermediciner kan sedan 2004 registreras endast via den s k centrala ansökningsproceduren, och besluten är bindande för samtliga medlemsländer inom EU.
Ett stort antal cancerläkemedel har godkänts de senaste åren. Flertalet av dem tillhör gruppen målsökande läkemedel (targeted drugs) – läkemedel som är riktade mot specifika målstrukturer.
I allmänhet uttrycks dessa målstrukturer i såväl normala celler som cancerceller. Den nya generationen av cancerläkemedel kommer därför inte att vara fria från biverkningar. Snarare tycks biverkningsgraden korrelera med hur effektivt medlet är mot cancersjukdomen.
Viktigt vid utveckling av dessa nya läkemedel är att relevanta undergrupper av patienter som har stor nytta av behandlingen kan identifieras. Detta kan ske med biopsianalyser av tumörernas egenskaper och hur medlen metaboliseras via nedärvda genetiska polymorfier.
Dessa strategier, syftande till skräddarsydd behandling, är en självklar och central del av modern läkemedelsutveckling.
Cancersjukdomarna har klassiskt behandlats med kirurgi och strålbehandling och sedan 1940-talet med ett växande antal läkemedel, främst cytostatika samt hormonellt verkande medel.
Under den senaste tioårsperioden har allt fler specifika läkemedel tillkommit som är riktade mot målstrukturer som anses ha stor betydelse för cancercellernas homeostas, proliferation och invasiva förmåga, s k målsökande läkemedel (targeted drugs). I allmänhet uttrycks dessa målstrukturer i såväl normala celler som cancerceller, men ibland med annan struktur och högre nivåer eller konstitutivt aktiverade i cancercellerna.
Denna nya generation av cancerläkemedel kommer i flertalet fall inte att vara helt selektiva och därför inte heller biverkningsfria. Snarare finns data som indikerar att graden av biverkningar tenderar att korrelera med effekten på cancersjukdomen.
Läkemedel får troligen en allt viktigare roll vid cancer
I en del fall är läkemedel den enda tillgängliga behandlingsformen. I första hand gäller det vid hematologiska maligniteter, men läkemedel har också haft en helt avgörande och botande roll vid exempelvis vissa typer av testikelcancer.
Läkemedlen ges ofta i kombination med kirurgi och/eller strålbehandling. Man har då ibland kunnat registrera tydligt förbättrad prognos, inklusive förbättrad överlevnad, speciellt om läkemedlen används som s k adjuvant behandling, dvs efter kirurgi/strålbehandling och i frånvaro av känd makroskopisk sjukdom.
För att belysa vikten av medicinsk behandling kan man ta bröstcancer som exempel. Adjuvant behandling med några kurer cytostatika kombinerat med tamoxifen, som i bröstcancerceller har en antagonistisk effekt på östrogenreceptorn, halverar efter 15 års uppföljning dödligheten i bröstcancer för den vanligaste typen, nämligen den som ses hos medelålders kvinnor med hormonkänslig cancer [1].
En förutsättning för framgångsrik cytostatikaanvändning är förbättrade stödjande och omvårdande insatser, som syftar till att minska vanligt förekommande biverkningar. Utan förbättrade antiemetikaregimer hade moderna onkologiska behandlingsprinciper inte kunnat tillämpas, och den polikliniska behandling som i dag är rutin skulle inte ha varit möjlig. Rutinmässigt används också granulocytkolonistimulerande faktorer (G-CSF) vid vissa mer dosintensiva regimer i syfte att reducera risken för svåra infektioner.
Läkemedel kommer sannolikt att få en allt viktigare roll vid behandling av patienter med tumörsjukdomar, både för att förbättra överlevnaden och för att fördröja sjukdomsprogress.
Kostnaden kan dock vara stor och vinsten modest för dessa centralt för hela EU registrerade läkemedel. Den hälsoekonomiska dimensionen värderas dock nationellt, i Sverige av Tandvårds- och läkemedelsförmånsverket (TLV), som också beslutar om ett registrerat (cancer)läkemedel ska omfattas av högkostnadsskyddet eller inte.
Trender för nya behandlingsstrategier och cancerläkemedel
Kliniska studier kommer i framtiden förhoppningsvis inte att vara fokuserade på att belägga små skillnader mellan ett nytt läkemedel och bästa tillgängliga standard i stora, biologiskt heterogena patientgrupper. Det är i flertalet fall varken hälsoekonomiskt eller tumörbiologiskt försvarbart. I stället bör utvecklingen av nya läkemedel redan på preklinisk nivå inriktas mot att identifiera subgrupper av tumörer, där målstrukturer uttrycks på sådant sätt att hög klinisk effekt kan förväntas.
För att den vidare kliniska utvecklingen ska vara framgångsrik krävs en förändring i strategierna för omhändertagandet av cancerpatienter. Färskfrusen cancervävnad med tillhörande normalvävnad behövs för analys av DNA-förändringar, genexpressionsprofiler, receptoruttryck och aktivering av signalvägar. Inom något år kommer det sannolikt också att vara möjligt att på lika detaljerat sätt analysera proteinuttrycket i cancerceller och normala celler. Till detta kommer att dosvalet för vissa läkemedel kan förfinas baserat på genetiska analyser.
Dessa gen- och proteinförändringar kommer rimligen att i många fall utgöra basen för val av rätt läkemedel till rätt patient; dagens histopatologiska indelning av diagnosgrupper kommer förhoppningsvis att kompletteras och modifieras av en mer funktionell indelning på basis av dessa kunskaper.
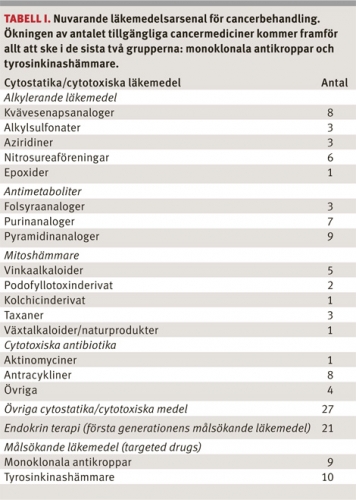
För att möjliggöra denna utveckling kommer det att behövas en förbättrad infrastruktur i sjukvården, med omhändertagande av normalvävnad och tumörvävnad enligt ovan, för att de drygt 100 cancermediciner som beräknas vara på marknaden år 2013 ska kunna användas på bästa sätt (Tabell I).
Registreringsprocedur för cancerläkemedel i Europa
Cancerläkemedel registrerades före Sveriges inträde i EU 1995 på nationell basis av Läkemedelsverket. Sedan dess har i praktiken nästan alla cancerläkemedel godkänts via den s k centrala ansökningsproceduren, även om ett fåtal medel godkänts baserat på registrering i andra EU-länder.
I den centrala proceduren värderar CHMP (Committee for Medicinal Products for Human Use), den europeiska läkemedelsnämnden, om nytta–riskförhållandet är positivt. Om så är fallet, godkänns sedan läkemedlet av EU-kommissionen i Bryssel. Sedan 2004 kan nya cancerläkemedel godkännas endast via den centrala proceduren.
Godkännandeprocessen inom EU
Hur cancermediciner och målsökande läkemedel bör utvecklas finns beskrivet i Riktlinjer för utveckling av cancerläkemedel [2], utgivna av europeiska läkemedelsmyndigheten EMEA (sedan december 2009 EMA). Där återfinns också hänvisningar till andra riktlinjer, som beskriver bl a principerna för biostatistik, farmakokinetiskt utvecklingsarbete och vilka prekliniska data som ska finnas, t ex innan den kliniska utvecklingen kan inledas.
När ansökan om registrering inkommit utser EMA två rapportörer (rapportör respektive medrapportör) ur kretsen av medlemmar i CHMP, i praktiken företrädare för två nationella läkemedelsmyndigheter. I valet av rapportör förväntas »kompetens« vara överordnat t ex »rättvis fördelning«. Sedan inträdet i EU har Läkemedelsverket varit den myndighet som näst flest gånger agerat som rapportör.
Rapportören respektive medrapportören sammanställer var sin rapport, som andra myndigheter samt läkemedelsföretaget har möjlighet att inkomma med kommentarer och synpunkter på. Rapportörerna tillställer företaget ett antal frågor, som måste besvaras på ett tillfredsställande sätt.
Om nytta–riskvärderingen bedömts som komplicerad eller kontroversiell eller om det föreligger intrikata vetenskapliga frågor, remitteras ärendet till CHMP:s vetenskapliga råd (Scientific Advisory Group, SAG) i onkologi och hematologi, bestående av nio europeiska onkologer/hematologer. CHMP tillställer SAG ett antal frågor, som man önskar få svar på. Detta sker genom att gruppen har ett sammanträde dit också läkemedelsföretaget kallas för en hearing. Baserat på den insända dokumentationen och vad som framkommer vid hearingen svarar sedan SAG på de frågor CHMP ställt.
CHMP ska, med eller utan hörande av SAG, sedan fatta ett majoritetsbeslut senast dag 210; »den regulatoriska klockan« kan dock vara stoppad i olika faser av handläggningen. Läkemedelsföretaget kallas ofta också till en hearing inför CHMP-gruppen. Beslut tas därefter genom omröstning bland de 27 representanterna för de nationella myndigheterna i EU. Island och Norge är också representerade men har ingen rösträtt. Det formella beslutet fattas sedan av EU-kommissionen. Processen från ansökan till beslut kan i komplexa ärenden ta längre tid, men i fall med en tydligt positiv nytta–riskbalans kan det också gå väsentligen snabbare.
Ett läkemedelsföretag som får avslag på en registreringsansökan har möjlighet att begära omprövning. Då utses nya rapportörer, och i praktiken konsulteras alltid SAG.
Nytänkande i den europeiska tillståndsprocessen
CHMP har alltså som uppgift att värdera om nytta–riskförhållandet är positivt eller inte. Värderingen omfattar i varje enskilt fall en balanserad analys av nyttan visavi biverkningarnas art, frekvens och svårighetsgrad. Detta ställs i relation till sjukdomens svårighetsgrad och tillgängligheten av alternativa läkemedel och andra behandlingsstrategier. Även om nytta–riskförhållandet värderats som positivt, kan det finnas brister i underlaget som gör att företaget åläggs att genomföra kompletterande studier efter godkännandet. Det kan avse utökat säkerhetsunderlag, men det kan också ha fokus på behandlingsprediktiva markörer och/eller optimering av doseringen av läkemedlet.
I detta sammanhang kan godkännandet av panitumumab nämnas som exempel på ett principiellt viktigt beslut. Denna monoklonala antikropp mot EGF(epidermal growth factor)-receptorn godkändes för behandling av kolorektal cancer endast i de fall tumören saknade aktiverande mutation i onkgenen KRAS. Beslutet baserade sig på ett antal icke-jämförande studier som genomförts med panitumumab och en annan monoklonal antikropp som också var riktad mot EGF-receptorn, cetuximab. Dessa studier indikerade frånvaro av tumörrespons vid muterad KRAS, något som senare bekräftades i en retrospektiv analys av den registreringsgrundande studien för panitumumab.
Detta har senare konfirmerats i retrospektiva analyser av ett antal randomiserade cetuximabstudier. Cetuximab fick därför i efterskott indikationerna för kolorektal cancer begränsade till icke-muterad KRAS.
Ett annat exempel är gefitinib vid icke-småcellig lungcancer. I de tidigaste studierna såg man bättre effekt vid adenokarcinom än vid skivepitelcancer hos icke-rökare, kvinnor och patienter med japanskt ursprung. Sedermera kunde man visa att individer med mutationer i EGF-receptorn hade betydligt högre sannolikhet för tumörrespons [3, 4]. Dessa och uppföljande studier visade att endast ungefär 10–15 procent av kaukasier med icke-småcellig lungcancer hade objektiv nytta av gefitinib och att effekten mycket starkt korrelerade med mutationer i EGF-receptorn. Gefitinib godkändes därför för behandling endast av subgruppen individer med icke-småcellig lungcancer med EGF-receptormutationer.
Viktigt identifiera behandlingsprediktiva markörer
Det är alltså av stor vikt att identifiera behandlingsprediktiva markörer för såväl gamla som nya cancerläkemedel. För detta behövs i princip tillgång till tumörvävnad och normal vävnad och undersökningar av typen PET (positronemissionstomografi) och magnetkamera, som kan avbilda funktionella skeenden och dynamiska förlopp över tid [2, 5].
Genom dessa strategier kommer man sannolikt att väsentligen kunna förbättra nytta–riskbalansen för den studerade medicinen. Dessa strategier kommer dock att ställa krav på validerade testsystem för de olika behandlingsprediktiva markörerna. Problemet är att vi för vissa av dessa målsökande mediciner saknar information om vilka mål som är funktionella. Ett exempel är njurcancer, där fem målsökande läkemedel är godkända, tyvärr utan att vi vet hur vi ska välja rätt medicin till rätt individ. Här behövs ytterligare studier enligt ovanstående principer för att kartlägga hur man bäst använder läkemedlen.
Analys av östrogen- och progesteronreceptorer och HER2/neu-uttryck hos alla bröstcancerpatienter, KRAS hos koloncancerpatienter, CD20 vid hematologiska maligniteter och EGF-receptormutationer hos individer med icke-småcellig lungcancer är exempel på tekniker som rutinmässigt används redan i dag i Sverige för att undvika överbehandling med mediciner som inte kommer att fungera om målstrukturer saknas.
Målsökande läkemedel kanske bör doseras till toxicitet
Eftersom de målstrukturer som finns uttryckta i cancercellerna i många fall även finns uttryckta i olika normalvävnader, är det inte orimligt att toxicitet skulle kunna fungera som »surrogatmarkör« för antitumoral effekt ur doseringssynvinkel [5]. I princip bör målsökande läkemedel doseras enligt principen om optimal biologisk dos. Medianöverlevnaden hos patienter som utvecklade markerad (grad 2+) hudrodnad vid behandling av icke-småcellig lungcancer med tyrosinkinashämmaren erlotinib var 11,1 månader jämfört med 3,3 månader hos individer med ingen/ringa hudtoxicitet [6]. En liknande effekt sågs också hos individer med pankreascancer [6].
I en liten, randomiserad studie sågs i en interimsanalys i numeriska tal en bättre effekt av cetuximab kombinerad med kemoterapi hos individer med kolorektal cancer som behandlades med eskalerande doser av cetuximab till hudtoxicitet jämfört med standarddosering [7].
Vid behandling med den monoklonala antikroppen bevacizumab, riktad mot VEGF (vascular endothelial growth factor), uppvisade individer som utvecklat ökat blodtryck på behandlingen möjligen en bättre antitumoral effekt [8].
I detta sammanhang bör också endokrin terapi för bröstcancerpatienter nämnas. I en retrospektiv analys av en randomiserad studie av adjuvant tamoxifen jämfört med aromatashämmaren anastrozol visades att individer som hade mer östrogenbristrelaterade biverkningar hade bättre effekt av båda dessa mediciner [9].
Motsvarande observationer finns också för konventionella cytostatika; individer utan nämnvärd toxicitet (mestadels är hematologisk toxicitet studerad) tenderade att ha sämre nytta av behandlingen [10]. Dessa skillnader mellan individer avseende registrerad toxicitet, trots att exempelvis cytostatikadosen kompenseras per kvadratmeter kroppsyta, skulle ju också i dessa fall kunna bero på genetiska polymorfier. Även detta område bör givetvis studeras vidare. Ett pragmatiskt sätt att kompensera för dessa interindividuella skillnader är att försöka att dosera behandlingen hos alla individer till samma grad av toxicitet.
Undantaget den randomiserade cetuximabstudien är ovanstående observationer svårtolkade, eftersom de rör sig om effekter som uppträder under pågående behandling. Därför kan »patientfaktorn« inte särskiljas från »behandlingsfaktorn« (jämför tumörrespons som prediktiv faktor för överlevnad). Dessa fynd bör därför ses som hypotesgenererande.
Oaktat denna viktiga invändning är det gemensamma temat för alla dessa observationer att cancermediciner, inkluderande målsökande läkemedel, sannolikt bör doseras mer individualiserat än vad som görs i dag, och då inte bara i form av dosreduktion vid toxicitet.
Framtida utvecklingsstrategier
I den framtida utvecklingen av cancermediciner kommer rimligen allt fler studier att genomföras, där man i detalj försöker kartlägga olika molekylära signaturer och hur dessa korrelerar till effekten av läkemedlen. Patienterna kommer inte nödvändigtvis att inkluderas i projekt på basis av histopatologisk diagnos, utan på basis av tumörens funktionella egenskaper.
För att studera tumörernas funktionella egenskaper krävs i dagsläget biopsianalyser. I vissa fall torde det vara möjligt att få information av denna karaktär också genom olika röntgenundersökningar, som förmår att visualisera olika funktionella egenskaper i tumören; i första hand PET-undersökningar respektive magnetkameraundersökningar med olika kontrastmedel.
I många sammanhang har det tagits för givet att primärtumören har i princip samma egenskaper som metastaserna. Det kan säkert vara så i vissa fall, men hos exempelvis individer med bröstcancer har man påvisat en påfallande brist på stabilitet i uttrycket av östrogen- och progesteronreceptorer och till delar också avseende onkgenen HER2/neu i jämförelse mellan primärtumör och motsvarande metastas. När målsökande läkemedel ska användas kan det alltså vara av stor vikt att man säkerställer att målstrukturerna för respektive läkemedel verkligen finns uttryckta på förväntat sätt även i metastassjukdomen.
Eftersom vi också börjar få en alltmer ökad kunskap om den interindividuella variationen av läkemedelsmetabolism, som till delar tycks vara styrd av genetiska polymorfier, kommer den framtida optimala doseringen av anticancermediciner inklusive målsökande läkemedel rimligtvis också att kräva denna information. Innan vi har denna detaljerade kunskap skulle en alternativ behandlingsmodell kunna vara att behandlingen doseras till en likvärdig grad av toxicitet hos varje individ. Detta kräver givetvis att det föreligger en påvisad korrelation mellan toxiciteten och registrerad effekt av behandling.
*
Potentiella bindningar eller jävsförhållanden: Jonas Bergh har varit medicinsk rådgivare åt ett stort antal läkemedels-, diagnostik- och konsultföretag; han har också varit ordförande/hållit föredrag och för detta erhållit arvode. Inga jäv i förhållande till innehållet i denna artikel.
Referenser
1. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687-717.
2. EMEA. Guideline on the evaluation of anticancer medicinal products in man. London: EMEA; 2006.
http://www.emea.europa.eu/pdfs/human/ewp/020595en.pdf
3. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from »never smokers« and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306-11.
4. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129-39.
5. Bergh J. Quo vadis with targeted drugs in the 21st century? J Clin Oncol. 2009;27(1):2-5.
6. Wacker B, Nagrani T, Weinberg J, Witt K, Clark G, Cagnoni PJ. Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin Cancer Res. 2007;13(13):3913-21.
7. Tejpar S, Peeters M, Humblet Y, Vermorken JB, De Hertogh G, De Roock W, et al, editors. Relationship of efficacy with KRAS status (wild type versus mutant) in patients with irinotecan-refractory metastatic colorectal cancer (mCRC), treated with irinotecan (q2w) and escalating doses of cetuximab (q1w): The EVEREST experience (preliminary data). 2008 ASCO Annual Meeting. J Clin Oncol. 2008;27(Suppl, pt I):169[abstract 4001].
8. Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A, et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol. 2008;26(28):4672-8.
9. Cuzick J, Sestak I, Cella D, Fallowfield L. Treatment-emergent endocrine symptoms and the risk of breast cancer recurrence: a retrospective analysis of the ATAC trial. Lancet Oncol. 2008;9(12):1143-8.
10. Rosendahl M, Ahlgren J, Andersen J, Bergh J, Blomquist C, Lidbrink E, et al. The risk of amenorrhoea after adjuvant chemotherapy for early stage breast cancer is related to inter-individual variations in chemotherapy-induced leukocyte nadir in young patients: data from the randomised SBG 2000-1 study. Eur J Cancer. 2009;45(18):3198-204.
Summary
A large number of cancer drugs have been approved during the last decade, the majority belonging to the group of targeted drugs. New cancer drugs can since 2004 only obtain market approval by a centralized European regulatory procedure and decisions are binding for all member states within the European Union. The targets for these drugs are frequently expressed both in normal/and cancer cells. The new generation of cancer drug will therefore not be deprived from side-effects, on the contrary, there are data indicating that the degree of some side effects tend to correlate with the anti-tumour effects. Targeted drugs have relatively frequently been demonstrated to have small/moderate effects when used for therapy of metastatic disease, however, important exceptions exist. Some of the drugs have shown clearly positive effects when used in the adjuvant setting, for therapy of micro-metastatic disease. It is important when developing these drugs to identify relevant subgroups of patients who may have large benefit from therapy. In order to be able to identify subgroups, analysis of tumour biological characteristics in biopsies are likely required, together with detailed knowledge of differences in metabolism, potentially related to difference in single nucleotide polymorphisms. These strategies are aiming at tailored therapy strategies which should be a core part of modern cancer drug development.
Jonas Bergh, Bertil Jonsson
Correspondence: Jonas Bergh, Radiumhemmet, Karolinska institutet/Karolinska universitetssjukhuset, SE-17176 Stockholm, Sweden jonas.bergh@ki.se